Tutorial
Using the neonOS Package to Check for Duplicates and Join Tables
Last Updated: Oct 3, 2022
NEON observational data are diverse in both content and in data structure. Generally, data are published in a set of data tables, each of which corresponds to a particular activity: for example, several data products include a field collection data table, a sample processing data table, and a table of laboratory analytical results.
Joining data tables
Depending on the analysis you want to carry out, there may be data in multiple tables that you want to bring together. For example, it is very common that species identifications are in a different table from chemical composition or pathogen status. For species-specific analyses, you would need to draw on multiple tables. There are a variety of ways to do this, but one of the simplest is to join the tables and create a single flat table.
The Quick Start Guides and Data Product User Guides provide information about the relationships between different data tables, and we recommend you consult these documents for the data products you are working with to gain the full picture. Quick Start Guides (QSGs) are included in the download package when you download data via the Data Portal, and can also be viewed on the Data Product Details page for each data product; for example, see the page for Plant foliar traits. Scroll down to the Documentation section to see the QSG. The Data Product User Guide is also available in the Documentation menu.
To join related tables, the neonOS package provides the
joinTableNEON() function, which checks the Table joining section of
the QSG, and uses the information there to join the tables if
possible. If the join isn't possible, or if it requires customized code,
the function will return an informative error, and usually refer you
to the QSG for more details.
Duplicates
One of the most common data entry errors that occurs in NEON OS data is
duplicate entry. NEON data entry applications and ingest validation rules
are designed to prevent duplicate entry where possible, but errors can't
be avoided completely. Consequently, NEON metadata for each OS data product
(the variables file) includes an indicator of which data fields, taken
together, should define a unique record. This combination of fields is
called the "primary key" for the data table. The neonOS function
removeDups() uses these metadata to identify duplicate records.
Depending on the content of the duplicate records, they may be resolved to
a single record or marked as unresolvable - see below for details.
In this tutorial, we will de-duplicate and then join two data tables in the Aquatic plant bryophyte chemical properties (DP1.20063.001) data product, using it as an example to demonstrate the operation of these two functions. Then we will take a look at Mosquitoes sampled from CO2 traps (DP1.10043.001), which contains more complicated table relationships, and see how to modify the code to work with those tables.
Objectives
After completing this activity, you will be able to:
- Identify and resolve duplicate data using the
neonOSpackage - Join NEON data tables using the
neonOSpackage
Things You’ll Need To Complete This Tutorial
You will need a version of R (4.0 or higher. This code may work with
earlier versions but it hasn't been tested) and, preferably, RStudio
loaded on your computer to complete this tutorial.
Install R Packages
-
neonUtilities:
install.packages("neonUtilities") -
neonOS:
install.packages("neonOS")
Additional Resources
- NEON Data Portal
- NEON Code Hub
Set Up R Environment and Download Data
First install and load the necessary packages.
# install packages. you can skip this step if
# the packages are already installed
install.packages("neonUtilities")
install.packages("neonOS")
install.packages("ggplot2")
# load packages
library(neonUtilities)
library(neonOS)
library(ggplot2)
We'll use aquatic plant chemistry (DP1.20063.001) as the example dataset. Use
the neonUtilities function loadByProduct() to download and read in the
data. If you're not familiar with the neonUtilities package and how to use
it to access NEON data, we recommend you follow the Download and Explore NEON Data
tutorial before proceeding with this one.
Here, we'll use the same subset of aquatic plant chemistry data as in the Download and Explore tutorial. Get the data from the Prairie Lake, Suggs Lake, and Toolik Lake sites, specifying the 2022 data release.
apchem <- loadByProduct(dpID="DP1.20063.001",
site=c("PRLA","SUGG","TOOK"),
package="expanded",
release="RELEASE-2022",
check.size=F)
Copy each of the downloaded tables into the R environment.
list2env(apchem, .GlobalEnv)
Identify and Resolve Duplicates
As noted above, duplicate data entry is a common error in human
data entry. removeDups() uses the definitional metadata in the
variables file to identify duplicates. It requires two inputs:
the data table, and the corresponding variables file.
apl_biomass <- removeDups(data=apl_biomass,
variables=variables_20063)
## No duplicated key values found!
There were no duplicates found in the apl_biomass data table.
A duplicateRecordQF quality flag has been added to the table,
and you can confirm that there were no duplicates by checking
the values of the flag.
unique(apl_biomass$duplicateRecordQF)
## [1] 0
All data have flag value = 0, indicating they are not duplicated.
Let's check the apl_plantExternalLabDataPerSample table.
apl_plantExternalLabDataPerSample <- removeDups(
data=apl_plantExternalLabDataPerSample,
variables=variables_20063)
10 duplicated key values found, representing 20 non-unique records. Attempting to resolve.
|==========================================================================================| 100%
2 resolveable duplicates merged into matching records
2 resolved records flagged with duplicateRecordQF=1
16 unresolveable duplicates flagged with duplicateRecordQF=2
The function output tells you there were 20 duplicates (out of 26108
total data records). Four of those duplicates were resolved, leaving two
records flagged as resolved duplicates, with duplicateRecordQF=1. The
remaining 16 couldn't be resolved, and were flagged with
duplicateRecordQF=2.
What does it mean for duplicates to be resolved? Some duplicates represent
identical data that have been entered multiple times, whereas some duplicates
have the same values in the primary key, but differ in data values.
removeDups() has a fairly narrow set of criteria for resolving to a single
record:
- If one data record has empty fields that are populated in the other record, the non-empty values are retained.
- If records are identical except for uid, remarks, and/or personnel (
identifiedBy,recordedBy, etc) fields, unique values are concatenated within the non-matching fields, separated by|(pipes).
Records that can be merged to a single record by these criteria are flagged
with duplicateRecordQF=1. Records with mis-matched data that can't be merged
are retained as-is and flagged with duplicateRecordQF=2.
Note that even in fully identical duplicates, the uid field (universal
identifier) will always be unique. Thus the uid field in merged records
will always contain the pipe-delimited set of uids of the original records.
What does this look like in practice? Let's look at the two resolved duplicates:
apl_plantExternalLabDataPerSample[which(
apl_plantExternalLabDataPerSample$duplicateRecordQF==1),]
## uid domainID siteID
## 55 cca9850e-165d-4cb7-9872-eff490c79ffa|1f5a5389-c18c-4fbf-81d6-9cd45e65f48a D03 SUGG
## 60 e6cab47f-427b-47a9-a281-cbfe7c101fc3|368e16bb-23d4-4596-b624-b338777bc9bc D03 SUGG
## namedLocation collectDate sampleID sampleCondition
## 55 SUGG.AOS.reach 2016-02-22 SUGG.20160222.UTFO.Q5 condition ok
## 60 SUGG.AOS.reach 2016-02-22 SUGG.20160222.UTFO.Q5 condition ok
## laboratoryName analysisDate analyzedBy sampleType replicate
## 55 Academy of Natural Sciences of Drexel University 2016-07-12 OKgcKejxXbI= CN 1
## 60 Academy of Natural Sciences of Drexel University 2016-07-12 OKgcKejxXbI= CN 1
## sampleVolumeFiltered filterSize percentFilterAnalyzed analyte analyteConcentration plantAlgaeLabUnits
## 55 NA 25 100 carbon 34.01 percent
## 60 NA 25 100 nitrogen 2.84 percent
## method externalRemarks publicationDate release duplicateRecordQF
## 55 <NA> <NA> 20211221T225348Z RELEASE-2022 1
## 60 <NA> <NA> 20211221T225348Z RELEASE-2022 1
You can see that both records have two pipe-delimited uids, and are flagged.
Let's look at the unresolveable duplicates:
apl_plantExternalLabDataPerSample[which(
apl_plantExternalLabDataPerSample$duplicateRecordQF==2),]
## uid domainID siteID namedLocation collectDate sampleID
## 1 5fa69a8b-d19e-40f1-84a8-e46ed08cdc59 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.LISP2.1
## 2 cebefd95-f4c7-4e60-9b79-93efd3f27691 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.LISP2.3
## 3 a4b6d931-b093-419e-bb69-0b684219405e D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.LISP2.1
## 4 f462390b-2d4a-4aec-92e1-b46a740ec40d D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.NULU.7
## 5 d4fa7f65-6b2b-46fa-9bca-529745970c56 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.NULU.7
## 6 ab43c466-b235-4f3d-9146-a1f81a91012d D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.LISP2.3
## 7 1140a4c2-139e-40dc-a14d-c2446e3a5664 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.NULU.9
## 8 5201dc04-da3d-4bc2-b2bf-668ad4a3ddb5 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.NULU.7
## 9 1e36cdeb-4e94-431d-8342-5ce5ff6ac6ae D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.NULU.9
## 10 b98069a4-74fb-40d8-8b98-7a5aedea7912 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.NULU.9
## 11 9633d367-bd10-4588-a6e2-cbe954327bf6 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.NULU.9
## 12 309ccbe3-d4a8-4bd3-b773-85aa98dd7627 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.LISP2.1
## 13 6d6b1da2-049f-439c-81c9-add482f6fab8 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.LISP2.1
## 14 841e51f4-dcc9-45fc-937b-871678ff16f2 D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.LISP2.3
## 15 3e04c3a1-7e2f-4349-a1c2-822d736fc2cf D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.NULU.7
## 16 431a4418-fdd0-4566-8791-d16ecfce76eb D03 SUGG SUGG.AOS.reach 2014-07-09 SUGG.20140709.LISP2.3
## sampleCondition laboratoryName analysisDate analyzedBy sampleType
## 1 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 2 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 3 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 4 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 5 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 6 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 7 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 8 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 9 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 10 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 11 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 12 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 13 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 14 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 15 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## 16 condition ok Academy of Natural Sciences of Drexel University 2015-02-05 OKgcKejxXbI= CN
## replicate sampleVolumeFiltered filterSize percentFilterAnalyzed analyte analyteConcentration
## 1 1 NA 25 100 carbon 38.22
## 2 1 NA 25 100 nitrogen 2.26
## 3 1 NA 25 100 nitrogen 1.98
## 4 1 NA 25 100 carbon 43.79
## 5 1 NA 25 100 nitrogen 3.16
## 6 1 NA 25 100 carbon 39.77
## 7 1 NA 25 100 carbon 43.26
## 8 1 NA 25 100 nitrogen 3.14
## 9 1 NA 25 100 nitrogen 2.94
## 10 1 NA 25 100 carbon 43.20
## 11 1 NA 25 100 nitrogen 3.00
## 12 1 NA 25 100 carbon 38.23
## 13 1 NA 25 100 nitrogen 2.02
## 14 1 NA 25 100 nitrogen 2.23
## 15 1 NA 25 100 carbon 43.93
## 16 1 NA 25 100 carbon 39.66
## plantAlgaeLabUnits method externalRemarks publicationDate release duplicateRecordQF
## 1 percent <NA> Limnobium spongia 20220110T211020Z RELEASE-2022 2
## 2 percent <NA> Limnobium spongia 20220110T211020Z RELEASE-2022 2
## 3 percent <NA> Limnobium spongia 20220110T211020Z RELEASE-2022 2
## 4 percent <NA> Nuphar Luteum 20220110T211020Z RELEASE-2022 2
## 5 percent <NA> Nuphar Luteum 20220110T211020Z RELEASE-2022 2
## 6 percent <NA> Limnobium spongia 20220110T211020Z RELEASE-2022 2
## 7 percent <NA> Nuphar Luteum 20220110T211020Z RELEASE-2022 2
## 8 percent <NA> Nuphar Luteum 20220110T211020Z RELEASE-2022 2
## 9 percent <NA> Nuphar Luteum 20220110T211020Z RELEASE-2022 2
## 10 percent <NA> Nuphar Luteum 20220110T211020Z RELEASE-2022 2
## 11 percent <NA> Nuphar Luteum 20220110T211020Z RELEASE-2022 2
## 12 percent <NA> Limnobium spongia 20220110T211020Z RELEASE-2022 2
## 13 percent <NA> Limnobium spongia 20220110T211020Z RELEASE-2022 2
## 14 percent <NA> Limnobium spongia 20220110T211020Z RELEASE-2022 2
## 15 percent <NA> Nuphar Luteum 20220110T211020Z RELEASE-2022 2
## 16 percent <NA> Limnobium spongia 20220110T211020Z RELEASE-2022 2
The key for this data table is the sample identifier and analyte, and here there are multiple records with the same sample identifier, both for carbon and nitrogen values. The most likely scenario is that these are unlabeled replicate analyses, i.e., the lab ran multiple analyses on the same samples for quality control purposes, but forgot to label them accordingly.
Now, how should you proceed with your analysis? Of course, that is up to
you, and depends on your analysis. Because these appear to be unlabeled
analytical replicates, I would probably average the analyte values, but
a decision like this can't be made automatically - removeDups() can
identify the records of concern, but what to do with them is a
judgement call.
Of course, NEON scientists also review NEON data and identify duplicates as part of quality assurance and quality control procedures, and resolve them if possible. In the data download step above, we accessed RELEASE-2022. The data release is stable and reproducible over time, but duplicates you find in one release may be resolved in future releases.
Join Data Tables
If you are using neonOS to check for duplicates and also to join data
tables, the duplicate step should always come first. Because duplicate
identification uses the variables file to determine uniqueness of data
records, the duplicate check step requires that the data match the
variables file exactly, which they won't after being joined.
To join the apl_biomass and apl_plantExternalLabDataPerSample tables,
we input both tables to the joinTableNEON() function. It uses the
information provided in NEON quick start guides to determine whether the
join is possible, and if it is, which fields to use to perform the join.
aqbc <- joinTableNEON(apl_biomass,
apl_plantExternalLabDataPerSample)
After joining tables, always take a look at the resulting table and make sure it makes sense. Errors in joining can easily result in completely nonsensical data. If you're not familiar with table joining operations, check out a lesson on the basics. The chapter on relational data in R for Data Science is a good one.
When checking your results, keep in mind that the default behavior of
joinTableNEON() is a full join, i.e., all records from both original
tables are retained, even if they don't match. For a small number of
table pairs, the Quick Start Guide specifies a left join, and in those
cases joinTableNEON() performs a left join.
Let's take a look at the aquatic plant table join:
nrow(apl_biomass)
## [1] 268
nrow(apl_plantExternalLabDataPerSample)
## [1] 572
nrow(aqbc)
## [1] 661
The number of rows in the joined table is larger than both of the original tables, but smaller than the sum of the original two. This suggests that most of the records in each of the original tables had a matching record in the other table, but some didn't.
View the full table:
View(aqbc)
(Table not displayed here due to size)
Here we can see we have several rows per chemSubsampleID for most
chemSubsampleIDs. Each row corresponds to one of the chemical
analytes, and the biomass data are repeated on each row. At the bottom
of the table are a number of biomass records with no corresponding
chemistry data; these explain why the merged table is larger than the
original chemistry table.
This table structure is consistent with the original tables and with the intended join, so we're satisfied all is well. If you're working with a different data product and encounter something unexpected or undesirable in a joined table, contact NEON using the Contact Us page.
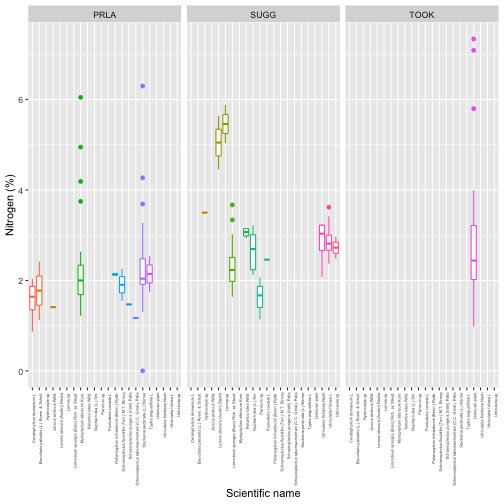
We can now connect chemical content to taxon, as in the Download and Explore NEON Data tutorial. Let's look at nitrogen content by species and site:
gg <- ggplot(subset(aqbc, analyte=="nitrogen"),
aes(scientificName, analyteConcentration,
group=scientificName,
color=scientificName)) +
geom_boxplot() +
facet_wrap(~siteID) +
theme(axis.text.x=element_text(angle=90,
hjust=1,
size=4)) +
theme(legend.position="none") +
ylab("Nitrogen (%)") +
xlab("Scientific name")
gg
Other Input Options
After downloading the data above, we ran list2env() to make each table
an independent object in the R environment, and then provided the tables
to the two functions as-is. This worked because the names of the objects
were identical to the table names, so the functions were able to figure
out which tables they were. If the object names are not exactly equal to
the table names, you will need to input the table names separately. If
we hadn't used list2env(), this is how we would proceed:
bio.dup <- removeDups(data=apchem$apl_biomass,
variables=apchem$variables_20063,
table="apl_biomass")
chem.dup <- removeDups(data=apchem$apl_plantExternalLabDataPerSample,
variables=apchem$variables_20063,
table="apl_plantExternalLabDataPerSample")
aq.join <- joinTableNEON(table1=bio.dup,
table2=chem.dup,
name1="apl_biomass",
name2="apl_plantExternalLabDataPerSample")
More Complicated Table Joins
In the aquatic plant chemistry example, we were able to join two tables
in a single step. In some cases, the relationship between tables is
more complicated, and joining is more difficult. In these cases,
joinTableNEON() will provide an error message, and usually direct
you to the Quick Start Guide for more information. Let's walk through
how you can use this information, using Mosquitoes sampled from CO2 traps
(DP1.10043.001) from Toolik Lake as an example.
mos <- loadByProduct(dpID="DP1.10043.001",
site="TOOL",
release="RELEASE-2022",
check.size=F)
list2env(mos, .GlobalEnv)
Let's say we're interested in evaluating which mosquito species are found in
which vegetation types. The species identifications are in the
mos_expertTaxonomistIDProcessed table, and the trapping conditions are in
the mos_trapping table. So we'll attempt to join those two tables.
mos.sp <- joinTableNEON(mos_trapping,
mos_expertTaxonomistIDProcessed)
Error in joinTableNEON(mos_trapping, mos_expertTaxonomistIDProcessed) :
Tables mos_trapping and mos_expertTaxonomistIDProcessed can't be joined directly, but can each be joined to a common table(s). Consult quick start guide for details.
The function returns an error, telling us it can't perform a simple join on the two tables, but there is a table they can each join to, which can be used to join them indirectly. As directed, we refer to the Quick Start Guide for DP1.10043.001, on the Data Product Details page.
From the QSG, we learn that mos_sorting is the intermediate table
between mos_trapping and mos_expertTaxonomistIDProcessed.
First, let's join mos_trapping and mos_sorting:
mos.trap <- joinTableNEON(mos_trapping,
mos_sorting)
Now, this next step is a bit odd. We've created a merged table of
mos_trapping and mos_sorting, but we know
mos_expertTaxonomistIDProcessed can only join to mos_sorting. So
we pass the merged table to joinTableNEON(), telling the function to
use the join instructions for mos_sorting and
mos_expertTaxonomistIDProcessed.
mos.tax <- joinTableNEON(mos.trap,
mos_expertTaxonomistIDProcessed,
name1="mos_sorting")
When you join data in this way, check carefully that the resulting table is structured logically and contains the data you expect it to. Looking at the merged table, we now have multiple records for each trapping event, with one record for each species captured in that event, plus a set of records for traps that either caught no mosquitoes or couldn't be deployed, and thus have no species identifications. This is consistent with the table join we performed, so everything appears to be correct.
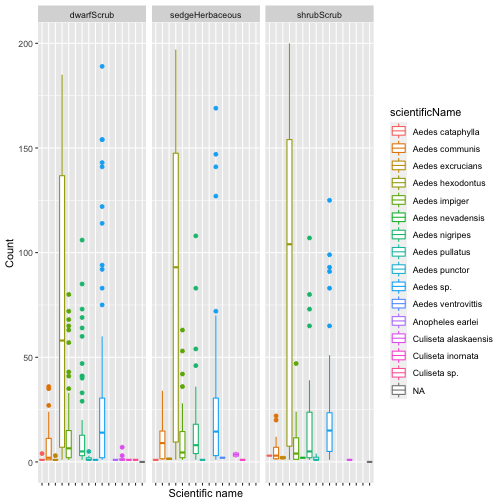
Let's take a look at species occurrence by habitat, as we set out to do:
gg <- ggplot(mos.tax,
aes(scientificName, individualCount,
group=scientificName,
color=scientificName)) +
geom_boxplot() +
facet_wrap(~nlcdClass) +
theme(axis.text.x=element_blank()) +
ylab("Count") +
xlab("Scientific name")
gg
## Warning: Removed 599 rows containing non-finite values (stat_boxplot).