This tutorial covers what metadata are, and why we need to work with
metadata. It covers the 3 most common metadata formats: text file format,
web page format and Ecological Metadata Language (EML).
R Skill Level: Introduction - you've got the basics of R down and
understand the general structure of tabular data.
Learning Objectives
After completing this tutorial, you will be able to:
Import a .csv file and examine the structure of the related R
object.
Use a metadata file to better understand the content of a dataset.
Explain the importance of including metadata details in your R script.
Describe what an EML file is.
Things You’ll Need To Complete This Tutorial
You will need the most current version of R and, preferably, RStudio loaded on
your computer to complete this tutorial.
Install R Packages
When presented in a workshop, the EML package will be presented as a
demonstration!
If completing EML portion of tutorial on your own, you must
install EML directly from GitHub (the package is in development and is not
yet on CRAN). You will need to install devtools to do this.
R Script & Challenge Code: NEON data lessons often contain challenges that reinforce
learned skills. If available, the code for challenge solutions is found in the
downloadable R script of the entire lesson, available in the footer of each lesson page.
Understand Our Data
In order to work with any data, we need to understand three things about the
data:
Structure
Format
Processing methods
If the data are collected by other people and organizations, we might also need
further information about:
What metrics are included in the dataset
The units those metrics were stored in
Explanation of where the metrics are stored in the data and what they are "called"
(e.g. what are the column names in a spreadsheet)
The time range that it covers
The spatial extent that the data covers
The above information, and more are stored in metadata - data
about the data. Metadata is information that describes a dataset and is integral
to working with external data (data that we did not collect ourselves).
Metadata Formats
Metadata come in different formats. We will discuss three of those in this tutorial:
Ecological Metadata Language (EML): A standardized metadata format stored
in xml format which is machine readable. Metadata has some standards however it's
common to encounter metadata stored differently in EML files created by different
organizations.
Text file: Sometimes metadata files can be found in text files that are either
downloaded with a data product OR that are available separately for the data.
Directly on a website (HTML / XML): Sometimes data are documented directly
in text format, on a web page.
**Data Tip:** When you find metadata for a dataset
that you are working with, immediately **DOWNLOAD AND SAVE IT** to the directory
on your computer where you saved the data. It is also a good idea to document
the URL where you found the metadata and data in a "readme" text file!
Metadata Stored on a Web Page
The metadata for the data that we are working with for the Harvard Forest field
site are stored in both EML format and on a web page. Let's explore the web
page first.
Let's begin by visiting that page above. At the top of the page, there is a list
of data available for Harvard Forest. NOTE: hf001-06: daily (metric) since
2001 (preview) is the data that we used in the
previous tutorial.
Scroll down to the Overview section on the website. Take note of the
information provided in that section and answer the questions in the
Challenge below.
### Challenge: Explore Metadata
Explore the metadata stored on the Harvard Forest LTER web page. Answer the
following questions.
What is the time span of the data available for this site?
You have some questions about these data. Who is the lead investigator / who
do you contact for more information? And how do you contact them?
Where is this field site located? How is the site location information stored
in the metadata? Is there any information that may be useful to you viewing the
location? HINT: What if you were not familiar with Harvard as a site / from
another country, etc?
Field Site Information: What is the elevation for the site? What is the
dominant vegetation cover for the site? HINT: Is dominant vegetation easy to
find in the metadata?
How is snow dealt with in the precipitation data?
Are there some metadata attributes that might be useful to access in a script
in R or Python rather than viewed on a web page? HINT: Can you answer all of
the questions above from the information provided on this website? Is there
additional information that you might want to find on the page?
View Metadata For Metrics of Interest
For this tutorial series, we are interested in the drivers of plant phenology -
specifically air and soil temperature, precipitation and photosynthetically
active radiation (PAR). Let's look for descriptions of these variables in the
metadata and determine several key attributes that we will need prior to working
with the data.
### Challenge: Metrics of Interest Metadata
View the metadata at the URL above. Answer the following questions about the
Harvard Forest LTER data - hf001-10: 15-minute (metric) since 2005:
What is the column heading name where each variable (air temperature, soil
temperature, precipitation and PAR) is stored?
What are the units that each variable are stored in?
What is the frequency of measurement collected for each and how are noData
values stored?
Where is the date information stored (in what field) and what timezone are
the dates stored in?
Why Metadata on a Web Page Is Not Ideal
It is nice to have a web page that displays metadata information, however
accessing metadata on a web page is difficult:
If the web page URL changes or the site goes down, the information is lost.
It's also more challenging to access metadata in text format on a web page
programatically - like using R as an interface - which we often
want to do when working with larger datasets.
A machine readable metadata file is better - especially when we are working with
large data and we want to automate and carefully document workflows. The
Ecological Metadata Language (EML) is one machine readable metadata format.
Ecological Metadata Language (EML)
While much of the documentation that we need to work with at the Harvard Forest
field site is available directly on the
Harvard Forest Data Archive page,
the website also offers metadata in EML format.
Introduction to EML
The Ecological Metadata Language (EML) is a data specification developed
specifically
to document ecological data. An EML file is created using a XML based format.
This means that content is embedded within hierarchical tags. For example,
the title of a dataset might be embedded in a <title> tag as follows:
<title>Fisher Meteorological Station at Harvard Forest since 2001</title>
Similarly, the creator of a dataset is also be found in a hierarchical tag
structure.
The EML package for R is designed to read and allow users to work with EML
formatted metadata. In this tutorial, we demonstrate how we can use EML in an
automated workflow.
NOTE: The EML package is still being developed, therefore we will not
explicitly teach all details of how to use it. Instead, we will provide
an example of how you can access EML files programmatically and background
information so that you can further explore EML and the EML package if you
need to work with it further.
EML Terminology
Let's first discuss some basic EML terminology. In the
context of EML, each EML file documents a dataset. This dataset may
consist of one or more files that contain the data in data tables. In the
case of our tabular meteorology data, the structure of our EML file includes:
The dataset. A dataset may contain
one or more data tables associated with it that may contain different types of
related information. For this Harvard Forest meteorological data, the data
tables contain tower measurements including precipitation and temperature, that
are aggregated at various time intervals (15 minute, daily, etc) and that date
back to 2001.
The data tables. Data tables refer to the actual data that make up the
dataset. For the Harvard Forest data, each data table contains a suite of
meteorological metrics, including precipitation and temperature (and associated
quality flags), that are aggregated at a particular time interval (e.g. one
data table contains monthly average data, another contains 15 minute
averaged data, etc).
Work With EML in R
To begin, we will load the EML package directly from ROpenSci's Git repository.
# install R EML tool
# load devtools (if you need to install "EML")
#library("devtools")
# IF YOU HAVE NOT DONE SO ALREADY: install EML from github -- package in
# development; not on CRAN
#install_github("ropensci/EML", build=FALSE, dependencies=c("DEPENDS", "IMPORTS"))
# load ROpenSci EML package
library(EML)
# load ggmap for mapping
library(ggmap)
# load tmaptools for mapping
library(tmaptools)
Next, we will read in the LTER EML file - directly from the online URL using
eml_read. This file documents multiple data products that can be downloaded.
Check out the
Harvard Forest Data Archive Page for Fisher Meteorological Station
for more on this dataset and to download the archive files directly.
Note that because this EML file is large, it takes a few seconds for the file to
load.
# data location
# http://harvardforest.fas.harvard.edu:8080/exist/apps/datasets/showData.html?id=hf001
# table 4 http://harvardforest.fas.harvard.edu/data/p00/hf001/hf001-04-monthly-m.csv
# import EML from Harvard Forest Met Data
# note, for this particular tutorial, we will work with an abridged version of the file
# that you can access directly on the harvard forest website. (see comment below)
eml_HARV <- read_eml("https://harvardforest1.fas.harvard.edu/sites/harvardforest.fas.harvard.edu/files/data/eml/hf001.xml")
# import a truncated version of the eml file for quicker demonstration
# eml_HARV <- read_eml("http://neonscience.github.io/NEON-R-Tabular-Time-Series/hf001-revised.xml")
# view size of object
object.size(eml_HARV)
## 1299568 bytes
# view the object class
class(eml_HARV)
## [1] "emld" "list"
The eml_read function creates an EML class object. This object can be
accessed using slots in R ($) rather than a typical subset [] approach.
Explore Metadata Attributes
We can begin to explore the contents of our EML file and associated data that it
describes. Let's start at the dataset level. We can use slots to view
the contact information for the dataset and a description of the methods.
# view the contact name listed in the file
eml_HARV$dataset$creator
## $individualName
## $individualName$givenName
## [1] "Emery"
##
## $individualName$surName
## [1] "Boose"
# view information about the methods used to collect the data as described in EML
eml_HARV$dataset$methods
## $methodStep
## $methodStep$description
## $methodStep$description$section
## $methodStep$description$section[[1]]
## [1] "<title>Observation periods</title><para>15-minute: 15 minutes, ending with given time. Hourly: 1 hour, ending with given time. Daily: 1 day, midnight to midnight. All times are Eastern Standard Time.</para>"
##
## $methodStep$description$section[[2]]
## [1] "<title>Instruments</title><para>Air temperature and relative humidity: Vaisala HMP45C (2.2m above ground). Precipitation: Met One 385 heated rain gage (top of gage 1.6m above ground). Global solar radiation: Licor LI200X pyranometer (2.7m above ground). PAR radiation: Licor LI190SB quantum sensor (2.7m above ground). Net radiation: Kipp and Zonen NR-LITE net radiometer (5.0m above ground). Barometric pressure: Vaisala CS105 barometer. Wind speed and direction: R.M. Young 05103 wind monitor (10m above ground). Soil temperature: Campbell 107 temperature probe (10cm below ground). Data logger: Campbell Scientific CR10X.</para>"
##
## $methodStep$description$section[[3]]
## [1] "<title>Instrument and flag notes</title><para>Air temperature. Daily air temperature is estimated from other stations as needed to complete record.</para><para>Precipitation. Daily precipitation is estimated from other stations as needed to complete record. Delayed melting of snow and ice (caused by problems with rain gage heater or heavy precipitation) is noted in log - daily values are corrected if necessary but 15-minute values are not. The gage may underestimate actual precipitation under windy or cold conditions.</para><para>Radiation. Whenever possible, snow and ice are removed from radiation instruments after precipitation events. Depth of snow or ice on instruments and time of removal are noted in log, but values are not corrected or flagged.</para><para>Wind speed and direction. During ice storms, values are flagged as questionable when there is evidence (from direct observation or the 15-minute record) that ice accumulation may have affected the instrument's operation.</para>"
Identify & Map Data Location
Looking at the coverage for our data, there is only one unique x and y value.
This suggests that our data were collected at (x,y) one point location. We know
this is a tower so a point location makes sense. Let's grab the x and y
coordinates and create a quick context map. We will use ggmap to create our
map.
NOTE: If this were a rectangular extent, we'd want the bounding box not just
the point. This is important if the data in raster, HDF5, or a similar format.
We need the extent to properly geolocate and process the data.
# grab x coordinate from the coverage information
XCoord <- eml_HARV$dataset$coverage$geographicCoverage$boundingCoordinates$westBoundingCoordinate[[1]]
# grab y coordinate from the coverage information
YCoord <- eml_HARV$dataset$coverage$geographicCoverage$boundingCoordinates$northBoundingCoordinate[[1]]
# make a map and add the xy coordinates to it
ggmap(get_stamenmap(rbind(as.numeric(paste(geocode_OSM("Massachusetts")$bbox))), zoom = 11, maptype=c("toner")), extent=TRUE) + geom_point(aes(x=as.numeric(XCoord),y=as.numeric(YCoord)),
color="darkred", size=6, pch=18)
The above example, demonstrated how we can extract information from an EML
document and use it programatically in R! This is just the beginning of what
we can do!
Metadata For Your Own Data
Now, imagine that you are working with someone else's data and you don't have a
metadata file associated with it? How do you know what units the data were in?
How the data were collected? The location that the data covers? It is equally
important to create metadata for your own data, to make your data more
efficiently "shareable".
This tutorial will demonstrate how to import a time series dataset stored in .csv
format into R. It will explore data classes for columns in a data.frame and
will walk through how to
convert a date, stored as a character string, into a date class that R can
recognize and plot efficiently.
Learning Objectives
After completing this tutorial, you will be able to:
Open a .csv file in R using read.csv()and understand why we
are using that file type.
Work with data stored in different columns within a data.frame in R.
Examine R object structures and data classes.
Convert dates, stored as a character class, into an R date
class.
Create a quick plot of a time-series dataset using qplot.
Things You’ll Need To Complete This Tutorial
You will need the most current version of R and, preferably, RStudio loaded on your computer to complete this tutorial.
R Script & Challenge Code: NEON data lessons often contain challenges that reinforce
learned skills. If available, the code for challenge solutions is found in the
downloadable R script of the entire lesson, available in the footer of each lesson page.
Data Related to Phenology
In this tutorial, we will explore atmospheric data (including temperature,
precipitation and other metrics) collected by sensors mounted on a
flux tower
at the NEON Harvard Forest field site. We are interested in exploring
changes in temperature, precipitation, Photosynthetically Active Radiation (PAR) and day
length throughout the year -- metrics that impact changes in the timing of plant
phenophases (phenology).
About .csv Format
The data that we will use is in .csv (comma-separated values) file format. The
.csv format is a plain text format, where each value in the dataset is
separate by a comma and each "row" in the dataset is separated by a line break.
Plain text formats are ideal for working both across platforms (Mac, PC, LINUX,
etc) and also can be read by many different tools. The plain text
format is also less likely to become obsolete over time.
To begin, let's import the data into R. We can use base R functionality
to import a .csv file. We will use the ggplot2 package to plot our data.
# Load packages required for entire script.
# library(PackageName) # purpose of package
library(ggplot2) # efficient, pretty plotting - required for qplot function
# set working directory to ensure R can find the file we wish to import
# provide the location for where you've unzipped the lesson data
wd <- "~/Git/data/"
**Data Tip:** Good coding practice -- install and
load all libraries at top of script.
If you decide you need another package later on in the script, return to this
area and add it. That way, with a glance, you can see all packages used in a
given script.
Once our working directory is set, we can import the file using read.csv().
# Load csv file of daily meteorological data from Harvard Forest
harMet.daily <- read.csv(
file=paste0(wd,"NEON-DS-Met-Time-Series/HARV/FisherTower-Met/hf001-06-daily-m.csv"),
stringsAsFactors = FALSE
)
stringsAsFactors=FALSE
When reading in files we most often use stringsAsFactors = FALSE. This
setting ensures that non-numeric data (strings) are not converted to
factors.
What Is A Factor?
A factor is similar to a category. However factors can be numerically interpreted
(they can have an order) and may have a level associated with them.
Examples of factors:
Month Names (an ordinal variable): Month names are non-numerical but we know
that April (month 4) comes after March (month 3) and each could be represented
by a number (4 & 3).
1 and 2s to represent male and female sex (a nominal variable): Numerical
interpretation of non-numerical data but no order to the levels.
After loading the data it is easy to convert any field that should be a factor by
using as.factor(). Therefore it is often best to read in a file with
stringsAsFactors = FALSE.
Data.Frames in R
The read.csv() imports our .csv into a data.frame object in R. data.frames
are ideal for working with tabular data - they are similar to a spreadsheet.
# what type of R object is our imported data?
class(harMet.daily)
## [1] "data.frame"
Data Structure
Once the data are imported, we can explore their structure. There are several
ways to examine the structure of a data frame:
head(): shows us the first 6 rows of the data (tail() shows the last 6
rows).
str() : displays the structure of the data as R interprets it.
Let's use both to explore our data.
# view first 6 rows of the dataframe
head(harMet.daily)
## date jd airt f.airt airtmax f.airtmax airtmin f.airtmin rh
## 1 2001-02-11 42 -10.7 -6.9 -15.1 40
## 2 2001-02-12 43 -9.8 -2.4 -17.4 45
## 3 2001-02-13 44 -2.0 5.7 -7.3 70
## 4 2001-02-14 45 -0.5 1.9 -5.7 78
## 5 2001-02-15 46 -0.4 2.4 -5.7 69
## 6 2001-02-16 47 -3.0 1.3 -9.0 82
## f.rh rhmax f.rhmax rhmin f.rhmin dewp f.dewp dewpmax f.dewpmax
## 1 58 22 -22.2 -16.8
## 2 85 14 -20.7 -9.2
## 3 100 34 -7.6 -4.6
## 4 100 59 -4.1 1.9
## 5 100 37 -6.0 2.0
## 6 100 46 -5.9 -0.4
## dewpmin f.dewpmin prec f.prec slrt f.slrt part f.part netr f.netr
## 1 -25.7 0.0 14.9 NA M NA M
## 2 -27.9 0.0 14.8 NA M NA M
## 3 -10.2 0.0 14.8 NA M NA M
## 4 -10.2 6.9 2.6 NA M NA M
## 5 -12.1 0.0 10.5 NA M NA M
## 6 -10.6 2.3 6.4 NA M NA M
## bar f.bar wspd f.wspd wres f.wres wdir f.wdir wdev f.wdev gspd
## 1 1025 3.3 2.9 287 27 15.4
## 2 1033 1.7 0.9 245 55 7.2
## 3 1024 1.7 0.9 278 53 9.6
## 4 1016 2.5 1.9 197 38 11.2
## 5 1010 1.6 1.2 300 40 12.7
## 6 1016 1.1 0.5 182 56 5.8
## f.gspd s10t f.s10t s10tmax f.s10tmax s10tmin f.s10tmin
## 1 NA M NA M NA M
## 2 NA M NA M NA M
## 3 NA M NA M NA M
## 4 NA M NA M NA M
## 5 NA M NA M NA M
## 6 NA M NA M NA M
# View the structure (str) of the data
str(harMet.daily)
## 'data.frame': 5345 obs. of 46 variables:
## $ date : chr "2001-02-11" "2001-02-12" "2001-02-13" "2001-02-14" ...
## $ jd : int 42 43 44 45 46 47 48 49 50 51 ...
## $ airt : num -10.7 -9.8 -2 -0.5 -0.4 -3 -4.5 -9.9 -4.5 3.2 ...
## $ f.airt : chr "" "" "" "" ...
## $ airtmax : num -6.9 -2.4 5.7 1.9 2.4 1.3 -0.7 -3.3 0.7 8.9 ...
## $ f.airtmax: chr "" "" "" "" ...
## $ airtmin : num -15.1 -17.4 -7.3 -5.7 -5.7 -9 -12.7 -17.1 -11.7 -1.3 ...
## $ f.airtmin: chr "" "" "" "" ...
## $ rh : int 40 45 70 78 69 82 66 51 57 62 ...
## $ f.rh : chr "" "" "" "" ...
## $ rhmax : int 58 85 100 100 100 100 100 71 81 78 ...
## $ f.rhmax : chr "" "" "" "" ...
## $ rhmin : int 22 14 34 59 37 46 30 34 37 42 ...
## $ f.rhmin : chr "" "" "" "" ...
## $ dewp : num -22.2 -20.7 -7.6 -4.1 -6 -5.9 -10.8 -18.5 -12 -3.5 ...
## $ f.dewp : chr "" "" "" "" ...
## $ dewpmax : num -16.8 -9.2 -4.6 1.9 2 -0.4 -0.7 -14.4 -4 0.6 ...
## $ f.dewpmax: chr "" "" "" "" ...
## $ dewpmin : num -25.7 -27.9 -10.2 -10.2 -12.1 -10.6 -25.4 -25 -16.5 -5.7 ...
## $ f.dewpmin: chr "" "" "" "" ...
## $ prec : num 0 0 0 6.9 0 2.3 0 0 0 0 ...
## $ f.prec : chr "" "" "" "" ...
## $ slrt : num 14.9 14.8 14.8 2.6 10.5 6.4 10.3 15.5 15 7.7 ...
## $ f.slrt : chr "" "" "" "" ...
## $ part : num NA NA NA NA NA NA NA NA NA NA ...
## $ f.part : chr "M" "M" "M" "M" ...
## $ netr : num NA NA NA NA NA NA NA NA NA NA ...
## $ f.netr : chr "M" "M" "M" "M" ...
## $ bar : int 1025 1033 1024 1016 1010 1016 1008 1022 1022 1017 ...
## $ f.bar : chr "" "" "" "" ...
## $ wspd : num 3.3 1.7 1.7 2.5 1.6 1.1 3.3 2 2.5 2 ...
## $ f.wspd : chr "" "" "" "" ...
## $ wres : num 2.9 0.9 0.9 1.9 1.2 0.5 3 1.9 2.1 1.8 ...
## $ f.wres : chr "" "" "" "" ...
## $ wdir : int 287 245 278 197 300 182 281 272 217 218 ...
## $ f.wdir : chr "" "" "" "" ...
## $ wdev : int 27 55 53 38 40 56 24 24 31 27 ...
## $ f.wdev : chr "" "" "" "" ...
## $ gspd : num 15.4 7.2 9.6 11.2 12.7 5.8 16.9 10.3 11.1 10.9 ...
## $ f.gspd : chr "" "" "" "" ...
## $ s10t : num NA NA NA NA NA NA NA NA NA NA ...
## $ f.s10t : chr "M" "M" "M" "M" ...
## $ s10tmax : num NA NA NA NA NA NA NA NA NA NA ...
## $ f.s10tmax: chr "M" "M" "M" "M" ...
## $ s10tmin : num NA NA NA NA NA NA NA NA NA NA ...
## $ f.s10tmin: chr "M" "M" "M" "M" ...
**Data Tip:** You can adjust the number of rows
returned when using the `head()` and `tail()` functions. For example you can use
`head(harMet.daily, 10)` to display the first 10 rows of your data rather than 6.
Classes in R
The structure results above let us know that the attributes in our data.frame
are stored as several different data types or classes as follows:
chr - Character: It holds strings that are composed of letters and
words. Character class data can not be interpreted numerically - that is to say
we can not perform math on these values even if they contain only numbers.
int - Integer: It holds numbers that are whole integers without decimals.
Mathematical operations can be performed on integers.
num - Numeric: It accepts data that are a wide variety of numeric formats
including decimals (floating point values) and integers. Numeric also accept
larger numbers than int will.
Storing variables using different classes is a strategic decision by R (and
other programming languages) that optimizes processing and storage. It allows:
data to be processed more quickly & efficiently.
the program (R) to minimize the storage size.
Differences Between Classes
Certain functions can be performed on certain data classes and not on others.
For example:
a <- "mouse"
b <- "sparrow"
class(a)
## [1] "character"
class(b)
## [1] "character"
# subtract a-b
a-b
## Error in a - b: non-numeric argument to binary operator
You can not subtract two character values given they are not numbers.
Additionally, performing summary statistics and other calculations of different
types of classes can yield different results.
# create a new object
speciesObserved <- c("speciesb","speciesc","speciesa")
speciesObserved
## [1] "speciesb" "speciesc" "speciesa"
# determine the class
class(speciesObserved)
## [1] "character"
# calculate the minimum
min(speciesObserved)
## [1] "speciesa"
# create numeric object
prec <- c(1,2,5,3,6)
# view class
class(prec)
## [1] "numeric"
# calculate min value
min(prec)
## [1] 1
We can calculate the minimum value for SpeciesObserved, a character
data class, however it does not return a quantitative minimum. It simply
looks for the first element, using alphabetical (rather than numeric) order.
Yet, we can calculate the quantitative minimum value for prec a numeric
data class.
Plot Data Using qplot()
Now that we've got classes down, let's plot one of the metrics in our data,
air temperature -- airt. Given this is a time series dataset, we want to plot
air temperature as it changes over time. We have a date-time column, date, so
let's use that as our x-axis variable and airt as our y-axis variable.
We will use the qplot() (for quick plot) function in the ggplot2 package.
The syntax for qplot() requires the x- and y-axis variables and then the R
object that the variables are stored in.
**Data Tip:** Add a title to the plot using
`main="Title string"`.
# quickly plot air temperature
qplot(x=date, y=airt,
data=harMet.daily,
main="Daily Air Temperature\nNEON Harvard Forest Field Site")
We have successfully plotted some data. However, what is happening on the
x-axis?
R is trying to plot EVERY date value in our data, on the x-axis. This makes it
hard to read. Why? Let's have a look at the class of the x-axis variable - date.
# View data class for each column that we wish to plot
class(harMet.daily$date)
## [1] "character"
class(harMet.daily$airt)
## [1] "numeric"
In this case, the date column is stored in our data.frame as a character
class. Because it is a character, R does not know how to plot the dates as a
continuous variable. Instead it tries to plot every date value as a text string.
The airt data class is numeric so that metric plots just fine.
Date as a Date-Time Class
We need to convert our date column, which is currently stored as a character
to a date-time class that can be displayed as a continuous variable. Lucky
for us, R has a date class. We can convert the date field to a date class
using as.Date().
# convert column to date class
harMet.daily$date <- as.Date(harMet.daily$date)
# view R class of data
class(harMet.daily$date)
## [1] "Date"
# view results
head(harMet.daily$date)
## [1] "2001-02-11" "2001-02-12" "2001-02-13" "2001-02-14" "2001-02-15"
## [6] "2001-02-16"
Now that we have adjusted the date, let's plot again. Notice that it plots
much more quickly now that R recognizes date as a date class. R can
aggregate ticks on the x-axis by year instead of trying to plot every day!
# quickly plot the data and include a title using main=""
# In title string we can use '\n' to force the string to break onto a new line
qplot(x=date,y=airt,
data=harMet.daily,
main="Daily Air Temperature w/ Date Assigned\nNEON Harvard Forest Field Site")
### Challenge: Using ggplot2's qplot function
Create a quick plot of the precipitation. Use the full time frame of data available
in the harMet.daily object.
Do precipitation and air temperature have similar annual patterns?
Create a quick plot examining the relationship between air temperature and precipitation.
Hint: you can modify the X and Y axis labels using xlab="label text" and
ylab="label text".
This tutorial defines Julian (year) day as most often used in an ecological
context, explains why Julian days are useful for analysis and plotting, and
teaches how to create a Julian day variable from a Date or Data/Time class
variable.
Learning Objectives
After completing this tutorial, you will be able to:
Define a Julian day (year day) as used in most ecological
contexts.
Convert a Date or Date/Time class variable to a Julian day
variable.
Things You’ll Need To Complete This Tutorial
You will need the most current version of R and, preferably, RStudio loaded on your computer to complete this tutorial.
R Script & Challenge Code: NEON data lessons often contain challenges that reinforce
learned skills. If available, the code for challenge solutions is found in the
downloadable R script of the entire lesson, available in the footer of each lesson page.
Convert Between Time Formats - Julian Days
Julian days, as most often used in an ecological context, is a continuous count
of the number of days beginning at Jan 1 each year. Thus each year will have up
to 365 (non-leap year) or 366 (leap year) days.
**Data Note:** This format can also be called ordinal
day or year day. In some contexts, Julian day can refer specifically to a
numeric day count since 1 January 4713 BCE or as a count from some other origin
day, instead of an annual count of 365 or 366 days.
Including a Julian day variable in your dataset can be very useful when
comparing data across years, when plotting data, and when matching your data
with other types of data that include Julian day.
Load the Data
Load this dataset that we will use to convert a date into a year day or Julian
day.
Notice the date is read in as a character and must first be converted to a Date
class.
# Load packages required for entire script
library(lubridate) #work with dates
# set working directory to ensure R can find the file we wish to import
wd <- "~/Git/data/"
# Load csv file of daily meteorological data from Harvard Forest
# Factors=FALSE so strings, series of letters/ words/ numerals, remain characters
harMet_DailyNoJD <- read.csv(
file=paste0(wd,"NEON-DS-Met-Time-Series/HARV/FisherTower-Met/hf001-06-daily-m-NoJD.csv"),
stringsAsFactors = FALSE
)
# what data class is the date column?
str(harMet_DailyNoJD$date)
## chr [1:5345] "2/11/01" "2/12/01" "2/13/01" "2/14/01" "2/15/01" ...
# convert "date" from chr to a Date class and specify current date format
harMet_DailyNoJD$date<- as.Date(harMet_DailyNoJD$date, "%m/%d/%y")
Convert with yday()
To quickly convert from from Date to Julian days, can we use yday(), a
function from the lubridate package.
# to learn more about it type
?yday
We want to create a new column in the existing data frame, titled julian, that
contains the Julian day data.
# convert with yday into a new column "julian"
harMet_DailyNoJD$julian <- yday(harMet_DailyNoJD$date)
# make sure it worked all the way through.
head(harMet_DailyNoJD$julian)
## [1] 42 43 44 45 46 47
tail(harMet_DailyNoJD$julian)
## [1] 268 269 270 271 272 273
**Data Tip:** In this tutorial we converted from
`Date` class to a Julian day, however, you can convert between any recognized
date/time class (POSIXct, POSIXlt, etc) and Julian day using `yday`.
This tutorial reviews how to plot a raster in R using the plot()
function. It also covers how to layer a raster on top of a hillshade to produce
an eloquent map.
Learning Objectives
After completing this tutorial, you will be able to:
Know how to plot a single band raster in R.
Know how to layer a raster dataset on top of a hillshade to create an elegant
basemap.
Things You’ll Need To Complete This Tutorial
You will need the most current version of R and, preferably, RStudio loaded
on your computer to complete this tutorial.
Set Working Directory: This lesson will explain how to set the working directory. You may wish to set your working directory to some other location, depending on how you prefer to organize your data.
In this tutorial, we will plot the Digital Surface Model (DSM) raster
for the NEON Harvard Forest Field Site. We will use the hist() function as a
tool to explore raster values. And render categorical plots, using the breaks
argument to get bins that are meaningful representations of our data.
We will use the terra package in this tutorial. If you do not
have the DSM_HARV variable as defined in the pervious tutorial, Intro To Raster In R, please download it using neonUtilities, as shown in the previous tutorial.
library(terra)
# set working directory
wd <- "~/data/"
setwd(wd)
# import raster into R
dsm_harv_file <- paste0(wd, "DP3.30024.001/neon-aop-products/2022/FullSite/D01/2022_HARV_7/L3/DiscreteLidar/DSMGtif/NEON_D01_HARV_DP3_732000_4713000_DSM.tif")
DSM_HARV <- rast(dsm_harv_file)
First, let's plot our Digital Surface Model object (DSM_HARV) using the
plot() function. We add a title using the argument main="title".
# Plot raster object
plot(DSM_HARV, main="Digital Surface Model - HARV")
Plotting Data Using Breaks
We can view our data "symbolized" or colored according to ranges of values
rather than using a continuous color ramp. This is comparable to a "classified"
map. However, to assign breaks, it is useful to first explore the distribution
of the data using a histogram. The breaks argument in the hist() function
tells R to use fewer or more breaks or bins.
If we name the histogram, we can also view counts for each bin and assigned
break values.
# Plot distribution of raster values
DSMhist<-hist(DSM_HARV,
breaks=3,
main="Histogram Digital Surface Model\n NEON Harvard Forest Field Site",
col="lightblue", # changes bin color
xlab= "Elevation (m)") # label the x-axis
# Where are breaks and how many pixels in each category?
DSMhist$breaks
## [1] 300 350 400 450
DSMhist$counts
## [1] 355269 611685 33046
Looking at our histogram, R has binned out the data as follows:
300-350m, 350-400m, 400-450m
We can determine that most of the pixel values fall in the 350-400m range with a
few pixels falling in the lower and higher range. We could specify different
breaks, if we wished to have a different distribution of pixels in each bin.
We can use those bins to plot our raster data. We will use the
terrain.colors() function to create a palette of 3 colors to use in our plot.
The breaks argument allows us to add breaks. To specify where the breaks
occur, we use the following syntax: breaks=c(value1,value2,value3).
We can include as few or many breaks as we'd like.
# plot using breaks.
plot(DSM_HARV,
breaks = c(300, 350, 400, 450),
col = terrain.colors(3),
main="Digital Surface Model (DSM) - HARV")
Data Tip: Note that when we assign break values
a set of 4 values will result in 3 bins of data.
Format Plot
If we need to create multiple plots using the same color palette, we can create
an R object (myCol) for the set of colors that we want to use. We can then
quickly change the palette across all plots by simply modifying the myCol
object.
We can label the x- and y-axes of our plot too using xlab and ylab.
# Assign color to a object for repeat use/ ease of changing
myCol = terrain.colors(3)
# Add axis labels
plot(DSM_HARV,
breaks = c(300, 350, 400, 450),
col = myCol,
main="Digital Surface Model - HARV",
xlab = "UTM Easting (m)",
ylab = "UTM Northing (m)")
Or we can also turn off the axes altogether.
# or we can turn off the axis altogether
plot(DSM_HARV,
breaks = c(300, 350, 400, 450),
col = myCol,
main="Digital Surface Model - HARV",
axes=FALSE)
Challenge: Plot Using Custom Breaks
Create a plot of the Harvard Forest Digital Surface Model (DSM) that has:
Six classified ranges of values (break points) that are evenly divided among
the range of pixel values.
Axis labels
A plot title
Hillshade & Layering Rasters
The terra package has built-in functions called terrain for calculating
slope and aspect, and shade for computing hillshade from the slope and aspect.
A hillshade is a raster that maps the shadows and texture that you would see
from above when viewing terrain.
The alpha value determines how transparent the colors will be (0 being
transparent, 1 being opaque). You can also change the color palette, for example,
use rainbow() instead of terrain.color().
For a full tutorial on rasters & raster data, please go through the
Intro to Raster Data in R tutorial
which provides a foundational concepts even if you aren't working with R.
A raster is a dataset made up of cells or pixels. Each pixel represents a value
associated with a region on the earth’s surface.
The spatial resolution of a raster refers the size of each cell
in meters. This size in turn relates to the area on the ground that the pixel
represents. Source: National Ecological Observatory Network
There are several ways that we can get from the point data collected by lidar to
the surface data that we want for different Digital Elevation Models or similar
data we use in analyses and mapping.
Basic gridding does not allow for the recovery/classification of data in any area
where data are missing. Interpolation (including Triangulated Irregular Network
(TIN) Interpolation) allows for gaps to be covered so that there are not holes
in the resulting raster surface.
Interpolation can be done in a number of different ways, some of which are
deterministic and some are probabilistic.
When converting a set of sample points to a grid, there are many
different approaches that should be considered. Source: National Ecological
Observatory Network
Gridding Points
When creating a raster, you may chose to perform a direct gridding of the data.
This means that you calculate one value for every cell in the raster where there
are sample points. This value may be a mean of all points, a max, min or some other
mathematical function. All other cells will then have no data values associated with
them. This means you may have gaps in your data if the point spacing is not well
distributed with at least one data point within the spatial coverage of each raster
cell.
When you directly grid a dataset, values will only be calculated
for cells that overlap with data points. Thus, data gaps will not be filled.
Source: National Ecological Observatory Network
We can create a raster from points through a process called gridding. Gridding is the process of taking a set of points and using them to create a surface composed of a regular grid.
Animation showing the general process of taking lidar point
clouds and converting them to a raster format. Source: Tristan Goulden,
National Ecological Observatory Network
Spatial Interpolation
Spatial interpolation involves calculating the value for a query point (or
a raster cell) with an unknown value from a set of known sample point values that
are distributed across an area. There is a general assumption that points closer
to the query point are more strongly related to that cell than those farther away.
However this general assumption is applied differently across different
interpolation functions.
Interpolation methods will estimate values for cells where no known values exist.
Deterministic & Probabilistic Interpolators
There are two main types of interpolation approaches:
Deterministic: create surfaces directly from measured points using a
weighted distance or area function.
Probabilistic (Geostatistical): utilize the statistical properties of the
measured points. Probabilistic techniques quantify the spatial auto-correlation
among measured points and account for the spatial configuration of the sample
points around the prediction location.
Different methods of interpolation are better for different datasets. This table
lays out the strengths of some of the more common interpolation methods.
We will focus on deterministic methods in this tutorial.
Deterministic Interpolation Methods
Let's look at a few different deterministic interpolation methods to understand
how different methods can affect an output raster.
Inverse Distance Weighted (IDW)
Inverse distance weighted (IDW) interpolation calculates the values of a query
point (a cell with an unknown value) using a linearly weighted combination of values
from nearby points.
IDW interpolation calculates the value of an unknown cell center value (a query point) using surrounding points with the assumption that closest points
will be the most similar to the value of interest. Source: QGIS
Key Attributes of IDW Interpolation
The raster is derived based upon an assumed linear relationship between the
location of interest and the distance to surrounding sample points. In other
words, sample points closest to the cell of interest are assumed to be more related
to its value than those further away.ID
Bounded/exact estimation, hence can not interpolate beyond the min/max range
of data point values. This estimate within the range of existing sample point
values can yield "flattened" peaks and valleys -- especially if the data didn't
capture those high and low points.
Interpolated points are average values
Good for point data that are equally distributed and dense. Assumes a consistent
trend or relationship between points and does not accommodate trends within the
data(e.g. east to west, elevational, etc).
IDW interpolation looks at the linear distance between the unknown value and surrounding points. Source: J. Abrecht, CUNY
Power
The power value changes the "weighting" of the IDW interpolation by specifying
how strongly points further away from the query point impact the calculated value
for that point. Power values range from 0-3+ with a default settings generally
being 2. A larger power value produces a more localized result - values further
away from the cell have less impact on it's calculated value, values closer to
the cell impact it's value more. A smaller power value produces a more averaged
result where sample points further away from the cell have a greater impact on
the cell's calculated value.
lower power values more averaged result, potential for a smoother surface.
As power decreases, the influence of sample points is larger. This yields a smoother
surface that is more averaged.
greater power values: more localized result, potential for more peaks and
valleys around sample point locations. As power increases, the influence of
sample points falls off more rapidly with distance. The query cell values become
more localized and less averaged.
IDW Take Home Points
IDW is good for:
Data whose distribution is strongly (and linearly) correlated with
distance. For example, noise falls off very predictably with distance.
Providing explicit control over the influence of distance (compared to Spline
or Kriging).
IDW is not so good for:
Data whose distribution depends on more complex sets of variables
because it can account only for the effects of distance.
Other features:
You can create a smoother surface by decreasing the power, increasing the
number of sample points used or increasing the search (sample points) radius.
You can create a surface that more closely represents the peaks and dips of
your sample points by decreasing the number of sample points used, decreasing
the search radius or increasing the power.
You can increase IDW surface accuracy by adding breaklines to the
interpolation process that serve as barriers. Breaklines represent abrupt
changes in elevation, such as cliffs.
Spline
Spline interpolation fits a curved surface through the sample points of your
dataset. Imagine stretching a rubber sheet across your points and gluing it to
each sample point along the way -- what you get is a smooth stretched sheet with
bumps and valleys. Unlike IDW, spline can estimate values above and below the
min and max values of your sample points. Thus it is good for estimating high
and low values not already represented in your data.
Estimating values outside of the range of sample input data.
Creating a smooth continuous surface.
Spline is not so good for:
Points that are close together and have large value differences. Slope calculations can yield over and underestimation.
Data with large, sudden changes in values that need to be represented (e.g., fault lines, extreme vertical topographic changes, etc). NOTE: some tools like ArcGIS have introduced a spline with barriers function in recent years.
Natural Neighbor Interpolation
Natural neighbor interpolation finds the closest subset of data points to the
query point of interest. It then applies weights to those points to calculate an
average estimated value based upon their proportionate areas derived from their
corresponding
Voronoi polygons
(see figure below for definition). The natural neighbor interpolator adapts
locally to the input data using points surrounding the query point of interest.
Thus there is no radius, number of points or other settings needed when using
this approach.
This interpolation method works equally well on regular and irregularly spaced data.
A Voronoi diagram is created by taking pairs of points that are close together and drawing a line that is equidistant between them and perpendicular to the line connecting them. Source: Wikipedia
Natural neighbor interpolation uses the area of each Voronoi polygon associated
with the surrounding points to derive a weight that is then used to calculate an
estimated value for the query point of interest.
To calculate the weighted area around a query point, a secondary Voronoi diagram
is created using the immediately neighboring points and mapped on top of the
original Voronoi diagram created using the known sample points (image below).
A secondary Voronoi diagram is created using the immediately neighboring points and mapped on top of the original Voronoi diagram created using the
known sample points to created a weighted Natural neighbor interpolated raster.
Image Source: ESRI
Data where spatial distribution is variable (and data that are equally distributed).
Categorical data.
Providing a smoother output raster.
Natural Neighbor Interpolation is not as good for:
Data where the interpolation needs to be spatially constrained (to a particular number of points of distance).
Data where sample points further away from or beyond the immediate "neighbor points" need to be considered in the estimation.
Other features:
Good as a local interpolator
Interpolated values fall within the range of values of the sample data
Surface passes through input samples; not above or below
Supports breaklines
Triangulated Irregular Network (TIN)
The Triangulated Irregular Network (TIN) is a vector based surface where sample
points (nodes) are connected by a series of edges creating a triangulated surface.
The TIN format remains the most true to the point distribution, density and
spacing of a dataset. It also may yield the largest file size!
A TIN creating from LiDAR data collected by the NEON AOP over
the NEON San Joaquin (SJER) field site.
For more on the TIN process see this information from ESRI:
Overview of TINs
Interpolation in R, GrassGIS, or QGIS
These additional resources point to tools and information for gridding in R, GRASS GIS and QGIS.
R functions
The packages and functions maybe useful when creating grids in R.
gstat::idw()
stats::loess()
akima::interp()
fields::Tps()
fields::splint()
spatial::surf.ls()
geospt::rbf()
QGIS tools
Check out the documentation on different interpolation plugins
Interpolation
These hyperspectral remote sensing data provide information on the National Ecological Observatory Network'sSan Joaquin Experimental Range (SJER) field site in March of 2021. The data used in this lesson is the 1km by 1km mosaic tile named NEON_D17_SJER_DP3_257000_4112000_reflectance.h5. If you already completed the previous lesson in this tutorial series, you do not need to download this data again. The entire SJER reflectance dataset can be accessed from the NEON Data Portal.
Set Working Directory: This lesson assumes that you have set your working directory to the location of the downloaded and unzipped data subsets.
For this tutorial, you should be comfortable reading data from a HDF5 file and have a general familiarity with hyperspectral data. If you aren't familiar with these steps already, we highly recommend you work through the Introduction to Working with Hyperspectral Data in HDF5 Format in R tutorial before moving on to this tutorial.
Everything on our planet reflects electromagnetic radiation from the Sun, and different types of land cover often have dramatically different reflectance properties across the spectrum. One of the most powerful aspects of the NEON Imaging Spectrometer (NIS, or hyperspectral sensor) is that it can accurately measure these reflectance properties at a very high spectral resolution. When you plot the reflectance values across the observed spectrum, you will see
that different land cover types (vegetation, pavement, bare soils, etc.) have distinct patterns in their reflectance values, a property that we call the
'spectral signature' of a particular land cover class.
In this tutorial, we will extract the reflectance values for all bands of a single pixel to plot a spectral signature for that pixel. In order to do this,
we need to pair the reflectance values for that pixel with the wavelength values of the bands that are represented in those measurements. We will also need to adjust the reflectance values by the scaling factor that is saved as an 'attribute' in the HDF5 file. First, let's start by defining the working
directory and reading in the example dataset.
# Call required packages
library(rhdf5)
library(plyr)
library(ggplot2)
library(neonUtilities)
wd <- "~/data/" #This will depend on your local environment
setwd(wd)
If you haven't already downloaded the hyperspectral data tile (in one of the previous tutorials in this series), you can use the neonUtilities function byTileAOP to download a single reflectance tile. You can run help(byTileAOP) to see more details on what the various inputs are. For this exercise, we'll specify the UTM Easting and Northing to be (257500, 4112500), which will download the tile with the lower left corner (257000, 4112000).
byTileAOP(dpID = 'DP3.30006.001',
site = 'SJER',
year = '2021',
easting = 257500,
northing = 4112500,
savepath = wd)
This file will be downloaded into a nested subdirectory under the ~/data folder (your working directory), inside a folder named DP3.30006.001 (the Data Product ID). The file should show up in this location: ~/data/DP3.30006.001/neon-aop-products/2021/FullSite/D17/2021_SJER_5/L3/Spectrometer/Reflectance/NEON_D17_SJER_DP3_257000_4112000_reflectance.h5.
Now we can read in the file and look at the contents using h5ls. You can move this file to a different location, but make sure to change the path accordingly.
Next, let's read in the wavelength center associated with each band in the HDF5 file. We will later match these with the reflectance values and show both in our final spectral signature plot.
# read in the wavelength information from the HDF5 file
wavelengths <- h5read(h5_file,"/SJER/Reflectance/Metadata/Spectral_Data/Wavelength")
Extract Z-dimension data slice
Next, we will extract all reflectance values for one pixel. This makes up the spectral signature or profile of the pixel. To do that, we'll use the h5read() function. Here we pick an arbitrary pixel at (100,35), and use the NULL value to select all bands from that location.
# extract all bands from a single pixel
aPixel <- h5read(h5_file,"/SJER/Reflectance/Reflectance_Data",index=list(NULL,100,35))
# The line above generates a vector of reflectance values.
# Next, we reshape the data and turn them into a dataframe
b <- adply(aPixel,c(1))
# create clean data frame
aPixeldf <- b[2]
# add wavelength data to matrix
aPixeldf$Wavelength <- wavelengths
head(aPixeldf)
## V1 Wavelength
## 1 206 381.6035
## 2 266 386.6132
## 3 274 391.6229
## 4 297 396.6327
## 5 236 401.6424
## 6 236 406.6522
Scale Factor
Then, we can pull the spatial attributes that we'll need to adjust the reflectance values. Often, large raster data contain floating point (values with decimals) information. However, floating point data consume more space (yield a larger file size) compared to integer values. Thus, to keep the file sizes smaller, the data will be scaled by a factor of 10, 100, 10000, etc. This scale factor will be noted in the data attributes.
After completing this tutorial, you will be able to:
Filter data, alone and combined with simple pattern matching grepl().
Use the group_by function in dplyr.
Use the summarise function in dplyr.
"Pipe" functions using dplyr syntax.
Things You’ll Need To Complete This Tutorial
You will need the most current version of R and, preferably, RStudio loaded
on your computer to complete this tutorial.
Install R Packages
neonUtilities: install.packages("neonUtilities") tools for working with
NEON data
dplyr: install.packages("dplyr") used for data manipulation
Intro to dplyr
When working with data frames in R, it is often useful to manipulate and
summarize data. The dplyr package in R offers one of the most comprehensive
group of functions to perform common manipulation tasks. In addition, the
dplyr functions are often of a simpler syntax than most other data
manipulation functions in R.
Elements of dplyr
There are several elements of dplyr that are unique to the library, and that
do very cool things!
Dplyr aims to provide a function for each basic verb of data manipulating, like:
filter() (and slice())
filter rows based on values in specified columns
arrange()
sort data by values in specified columns
select() (and rename())
view and work with data from only specified columns
distinct()
view and work with only unique values from specified columns
mutate() (and transmute())
add new data to the data frame
summarise()
calculate specified summary statistics on data
sample_n() and sample_frac()
return a random sample of rows
Format of function calls
The single table verb functions share these features:
The first argument is a data.frame (or a dplyr special class tbl_df, known
as a 'tibble').
dplyr can work with data.frames as is, but if you're dealing with large
data it's worthwhile to convert them to a tibble, to avoid printing
a lot of data to the screen. You can do this with the function as_tibble().
Calling the class function on a tibble will return the vector
c("tbl_df", "tbl", "data.frame").
The subsequent arguments describe how to manipulate the data (e.g., based on
which columns, using which summary statistics), and you can refer to columns
directly (without using $).
The result is always a new tibble.
Function calls do not generate 'side-effects'; you always have to assign the
results to an object
Grouped operations
Certain functions (e.g., group_by, summarise, and other 'aggregate functions')
allow you to get information for groups of data, in one fell swoop. This is like
performing database functions with knowing SQL or any other db specific code.
Powerful stuff!
Piping
We often need to get a subset of data using one function, and then use
another function to do something with that subset (and we may do this multiple
times). This leads to nesting functions, which can get messy and hard to keep
track of. Enter 'piping', dplyr's way of feeding the output of one function into
another, and so on, without the hassleof parentheses and brackets.
Let's say we want to start with the data frame my_data, apply function1(),
then function2(), and then function3(). This is what that looks like without
piping:
function3(function2(function1(my_data)))
This is messy, difficult to read, and the reverse of the order our functions
actually occur. If any of these functions needed additional arguments, the
readability would be even worse!
The piping operator %>% takes everything in front of it and "pipes" it into
the first argument of the function after. So now our example looks like this:
This runs identically to the original nested version!
For example, if we want to find the mean body weight of male mice, we'd do this:
myMammalData %>% # start with a data frame
filter(sex=='M') %>% # first filter for rows where sex is male
summarise (mean_weight = mean(weight)) # find the mean of the weight
# column, store as mean_weight
This is read as "from data frame myMammalData, select only males and return
the mean weight as a new list mean_weight".
Download Small Mammal Data
Before we get started, we will need to download our data to investigate. To
do so, we will use the loadByProduct() function from the neonUtilities
package to download data straight from the NEON servers. To learn more about
this function, please see the Download and Explore NEON data tutorial here.
Let's look at the NEON small mammal capture data from Harvard Forest (within
Domain 01) for all of 2014. This site is located in the heart of the Lyme
disease epidemic.
# load packages
library(dplyr)
library(neonUtilities)
# load the NEON small mammal capture data
# NOTE: the check.size = TRUE argument means the function
# will require confirmation from you that you want to load
# the quantity of data requested
loadData <- loadByProduct(dpID="DP1.10072.001", site = "HARV",
startdate = "2014-01", enddate = "2014-12",
check.size = TRUE) # Console requires your response!
# if you'd like, check out the data
str(loadData)
The loadByProduct() function calls the NEON server, downloads the monthly
data files, and 'stacks' them together to form two tables of data called
'mam_pertrapnight' and 'mam_perplotnight'. It also saves the readme file,
explanations of variables, and validation metadata, and combines these all into
a single 'list' that we called 'loadData'. The only part of this list that we
really need for this tutorial is the 'mam_pertrapnight' table, so let's extract
just that one and call it 'myData'.
myData <- loadData$mam_pertrapnight
class(myData) # Confirm that 'myData' is a data.frame
## [1] "data.frame"
Use dplyr
For the rest of this tutorial, we are only going to be working with three
variables within 'myData':
scientificName a string of "Genus species"
sex a string with "F", "M", or "U"
identificationQualifier a string noting uncertainty in the species
identification
filter()
This function:
extracts only a subset of rows from a data frame according to specified
conditions
is similar to the base function subset(), but with simpler syntax
inputs: data object, any number of conditional statements on the named columns
of the data object
output: a data object of the same class as the input object (e.g.,
data.frame in, data.frame out) with only those rows that meet the conditions
For example, let's create a new dataframe that contains only female Peromyscus
mainculatus, one of the key small mammal players in the life cycle of Lyme
disease-causing bacterium.
# filter on `scientificName` is Peromyscus maniculatus and `sex` is female.
# two equals signs (==) signifies "is"
data_PeroManicFemales <- filter(myData,
scientificName == 'Peromyscus maniculatus',
sex == 'F')
# Note how we were able to put multiple conditions into the filter statement,
# pretty cool!
So we have a dataframe with our female P. mainculatus but how many are there?
# how many female P. maniculatus are in the dataset
# would could simply count the number of rows in the new dataset
nrow(data_PeroManicFemales)
## [1] 98
# or we could write is as a sentence
print(paste('In 2014, NEON technicians captured',
nrow(data_PeroManicFemales),
'female Peromyscus maniculatus at Harvard Forest.',
sep = ' '))
## [1] "In 2014, NEON technicians captured 98 female Peromyscus maniculatus at Harvard Forest."
That's awesome that we can quickly and easily count the number of female
Peromyscus maniculatus that were captured at Harvard Forest in 2014. However,
there is a slight problem. There are two Peromyscus species that are common
in Harvard Forest: Peromyscus maniculatus (deer mouse) and Peromyscus leucopus
(white-footed mouse). These species are difficult to distinguish in the field,
leading to some uncertainty in the identification, which is noted in the
'identificationQualifier' data field by the term "cf. species" or "aff. species".
(For more information on these terms and 'open nomenclature' please see this great Wiki article here).
When the field technician is certain of their identification (or if they forget
to note their uncertainty), identificationQualifier will be NA. Let's run the
same code as above, but this time specifying that we want only those observations
that are unambiguous identifications.
# filter on `scientificName` is Peromyscus maniculatus and `sex` is female.
# two equals signs (==) signifies "is"
data_PeroManicFemalesCertain <- filter(myData,
scientificName == 'Peromyscus maniculatus',
sex == 'F',
identificationQualifier =="NA")
# Count the number of un-ambiguous identifications
nrow(data_PeroManicFemalesCertain)
## [1] 0
Woah! So every single observation of a Peromyscus maniculatus had some level
of uncertainty that the individual may actually be Peromyscus leucopus. This
is understandable given the difficulty of field identification for these species.
Given this challenge, it will be best for us to consider these mice at the genus
level. We can accomplish this by searching for only the genus name in the
'scientificName' field using the grepl() function.
grepl()
This is a function in the base package (e.g., it isn't part of dplyr) that is
part of the suite of Regular Expressions functions. grepl uses regular
expressions to match patterns in character strings. Regular expressions offer
very powerful and useful tricks for data manipulation. They can be complicated
and therefore are a challenge to learn, but well worth it!
Here, we present a very simple example.
inputs: pattern to match, character vector to search for a match
output: a logical vector indicating whether the pattern was found within
each element of the input character vector
Let's use grepl to learn more about our possible disease vectors. In reality,
all species of Peromyscus are viable players in Lyme disease transmission, and
they are difficult to distinguise in a field setting, so we really should be
looking at all species of Peromyscus. Since we don't have genera split out as
a separate field, we have to search within the scientificName string for the
genus -- this is a simple example of pattern matching.
We can use the dplyr function filter() in combination with the base function
grepl() to accomplish this.
# combine filter & grepl to get all Peromyscus (a part of the
# scientificName string)
data_PeroFemales <- filter(myData,
grepl('Peromyscus', scientificName),
sex == 'F')
# how many female Peromyscus are in the dataset
print(paste('In 2014, NEON technicians captured',
nrow(data_PeroFemales),
'female Peromyscus spp. at Harvard Forest.',
sep = ' '))
## [1] "In 2014, NEON technicians captured 558 female Peromyscus spp. at Harvard Forest."
group_by() + summarise()
An alternative to using the filter function to subset the data (and make a new
data object in the process), is to calculate summary statistics based on some
grouping factor. We'll use group_by(), which does basically the same thing as
SQL or other tools for interacting with relational databases. For those
unfamiliar with SQL, no worries - dplyr provides lots of additional
functionality for working with databases (local and remote) that does not
require knowledge of SQL. How handy!
The group_by() function in dplyr allows you to perform functions on a subset
of a dataset without having to create multiple new objects or construct for()
loops. The combination of group_by() and summarise() are great for
generating simple summaries (counts, sums) of grouped data.
NOTE: Be continentious about using summarise with other summary functions! You
need to think about weighting for means and variances, and summarize doesn't
work precisely for medians if there is any missing data (e.g., if there was no
value recorded, maybe it was for a good reason!).
Continuing with our small mammal data, since the diversity of the entire small
mammal community has been shown to impact disease dynamics among the key
reservoir species, we really want to know more about the demographics of the
whole community. We can quickly generate counts by species and sex in 2 lines of
code, using group_by and summarise.
# how many of each species & sex were there?
# step 1: group by species & sex
dataBySpSex <- group_by(myData, scientificName, sex)
# step 2: summarize the number of individuals of each using the new df
countsBySpSex <- summarise(dataBySpSex, n_individuals = n())
## `summarise()` regrouping output by 'scientificName' (override with `.groups` argument)
# view the data (just top 10 rows)
head(countsBySpSex, 10)
## # A tibble: 10 x 3
## # Groups: scientificName [5]
## scientificName sex n_individuals
## <chr> <chr> <int>
## 1 Blarina brevicauda F 50
## 2 Blarina brevicauda M 8
## 3 Blarina brevicauda U 22
## 4 Blarina brevicauda <NA> 19
## 5 Glaucomys volans M 1
## 6 Mammalia sp. U 1
## 7 Mammalia sp. <NA> 1
## 8 Microtus pennsylvanicus F 2
## 9 Myodes gapperi F 103
## 10 Myodes gapperi M 99
Note: the output of step 1 (dataBySpSex) does not look any different than the
original dataframe (myData), but the application of subsequent functions (e.g.,
summarise) to this new dataframe will produce distinctly different results than
if you tried the same operations on the original. Try it if you want to see the
difference! This is because the group_by() function converted dataBySpSex
into a "grouped_df" rather than just a "data.frame". In order to confirm this,
try using the class() function on both myData and dataBySpSex. You can
also read the help documentation for this function by running the code:
?group_by()
# View class of 'myData' object
class(myData)
## [1] "data.frame"
# View class of 'dataBySpSex' object
class(dataBySpSex)
## [1] "grouped_df" "tbl_df" "tbl" "data.frame"
# View help file for group_by() function
?group_by()
Pipe functions together
We created multiple new data objects during our explorations of dplyr
functions, above. While this works, we can produce the same results more
efficiently by chaining functions together and creating only one new data object
that encapsulates all of the previously sought information: filter on only
females, grepl to get only Peromyscus spp., group_by individual species, and
summarise the numbers of individuals.
# combine several functions to get a summary of the numbers of individuals of
# female Peromyscus species in our dataset.
# remember %>% are "pipes" that allow us to pass information from one function
# to the next.
dataBySpFem <- myData %>%
filter(grepl('Peromyscus', scientificName), sex == "F") %>%
group_by(scientificName) %>%
summarise(n_individuals = n())
## `summarise()` ungrouping output (override with `.groups` argument)
# view the data
dataBySpFem
## # A tibble: 3 x 2
## scientificName n_individuals
## <chr> <int>
## 1 Peromyscus leucopus 455
## 2 Peromyscus maniculatus 98
## 3 Peromyscus sp. 5
Cool!
Base R only
So that is nice, but we had to install a new package dplyr. You might ask,
"Is it really worth it to learn new commands if I can do this is base R." While
we think "yes", why don't you see for yourself. Here is the base R code needed
to accomplish the same task.
# For reference, the same output but using R's base functions
# First, subset the data to only female Peromyscus
dataFemPero <- myData[myData$sex == 'F' &
grepl('Peromyscus', myData$scientificName), ]
# Option 1 --------------------------------
# Use aggregate and then rename columns
dataBySpFem_agg <-aggregate(dataFemPero$sex ~ dataFemPero$scientificName,
data = dataFemPero, FUN = length)
names(dataBySpFem_agg) <- c('scientificName', 'n_individuals')
# view output
dataBySpFem_agg
## scientificName n_individuals
## 1 Peromyscus leucopus 455
## 2 Peromyscus maniculatus 98
## 3 Peromyscus sp. 5
# Option 2 --------------------------------------------------------
# Do it by hand
# Get the unique scientificNames in the subset
sppInDF <- unique(dataFemPero$scientificName[!is.na(dataFemPero$scientificName)])
# Use a loop to calculate the numbers of individuals of each species
sciName <- vector(); numInd <- vector()
for (i in sppInDF) {
sciName <- c(sciName, i)
numInd <- c(numInd, length(which(dataFemPero$scientificName==i)))
}
#Create the desired output data frame
dataBySpFem_byHand <- data.frame('scientificName'=sciName,
'n_individuals'=numInd)
# view output
dataBySpFem_byHand
## scientificName n_individuals
## 1 Peromyscus leucopus 455
## 2 Peromyscus maniculatus 98
## 3 Peromyscus sp. 5
R Script & Challenge Code: NEON data lessons often contain challenges that reinforce
learned skills. If available, the code for challenge solutions is found in the
downloadable R script of the entire lesson, available in the footer of each lesson page.
Additional Resources
Consider reviewing the documentation for the
RHDF5 package.
About HDF5
The HDF5 file can store large, heterogeneous datasets that include metadata. It
also supports efficient data slicing, or extraction of particular subsets of a
dataset which means that you don't have to read large files read into the
computers memory / RAM in their entirety in order work with them.
To access HDF5 files in R, we will use the rhdf5 library which is part of
the Bioconductor
suite of R libraries. It might also be useful to install
the
free HDF5 viewer
which will allow you to explore the contents of an HDF5 file using a graphic interface.
First, let's get R setup. We will use the rhdf5 library. To access HDF5 files in
R, we will use the rhdf5 library which is part of the Bioconductor suite of R
packages. As of May 2020 this package was not yet on CRAN.
# Install rhdf5 package (only need to run if not already installed)
#install.packages("BiocManager")
#BiocManager::install("rhdf5")
# Call the R HDF5 Library
library("rhdf5")
# set working directory to ensure R can find the file we wish to import and where
# we want to save our files
wd <- "~/Git/data/" #This will depend on your local environment
setwd(wd)
Now, let's create a basic H5 file with one group and one dataset in it.
# Create hdf5 file
h5createFile("vegData.h5")
## [1] TRUE
# create a group called aNEONSite within the H5 file
h5createGroup("vegData.h5", "aNEONSite")
## [1] TRUE
# view the structure of the h5 we've created
h5ls("vegData.h5")
## group name otype dclass dim
## 0 / aNEONSite H5I_GROUP
Next, let's create some dummy data to add to our H5 file.
# create some sample, numeric data
a <- rnorm(n=40, m=1, sd=1)
someData <- matrix(a,nrow=20,ncol=2)
Write the sample data to the H5 file.
# add some sample data to the H5 file located in the aNEONSite group
# we'll call the dataset "temperature"
h5write(someData, file = "vegData.h5", name="aNEONSite/temperature")
# let's check out the H5 structure again
h5ls("vegData.h5")
## group name otype dclass dim
## 0 / aNEONSite H5I_GROUP
## 1 /aNEONSite temperature H5I_DATASET FLOAT 20 x 2
View a "dump" of the entire HDF5 file. NOTE: use this command with CAUTION if you
are working with larger datasets!
# we can look at everything too
# but be cautious using this command!
h5dump("vegData.h5")
## $aNEONSite
## $aNEONSite$temperature
## [,1] [,2]
## [1,] 0.33155432 2.4054446
## [2,] 1.14305151 1.3329978
## [3,] -0.57253964 0.5915846
## [4,] 2.82950139 0.4669748
## [5,] 0.47549005 1.5871517
## [6,] -0.04144519 1.9470377
## [7,] 0.63300177 1.9532294
## [8,] -0.08666231 0.6942748
## [9,] -0.90739256 3.7809783
## [10,] 1.84223101 1.3364965
## [11,] 2.04727590 1.8736805
## [12,] 0.33825921 3.4941913
## [13,] 1.80738042 0.5766373
## [14,] 1.26130759 2.2307994
## [15,] 0.52882731 1.6021497
## [16,] 1.59861449 0.8514808
## [17,] 1.42037674 1.0989390
## [18,] -0.65366487 2.5783750
## [19,] 1.74865593 1.6069304
## [20,] -0.38986048 -1.9471878
# Close the file. This is good practice.
H5close()
Add Metadata (attributes)
Let's add some metadata (called attributes in HDF5 land) to our dummy temperature
data. First, open up the file.
# open the file, create a class
fid <- H5Fopen("vegData.h5")
# open up the dataset to add attributes to, as a class
did <- H5Dopen(fid, "aNEONSite/temperature")
# Provide the NAME and the ATTR (what the attribute says) for the attribute.
h5writeAttribute(did, attr="Here is a description of the data",
name="Description")
h5writeAttribute(did, attr="Meters",
name="Units")
Now we can add some attributes to the file.
# let's add some attributes to the group
did2 <- H5Gopen(fid, "aNEONSite/")
h5writeAttribute(did2, attr="San Joaquin Experimental Range",
name="SiteName")
h5writeAttribute(did2, attr="Southern California",
name="Location")
# close the files, groups and the dataset when you're done writing to them!
H5Dclose(did)
H5Gclose(did2)
H5Fclose(fid)
Working with an HDF5 File in R
Now that we've created our H5 file, let's use it! First, let's have a look at
the attributes of the dataset and group in the file.
# look at the attributes of the precip_data dataset
h5readAttributes(file = "vegData.h5",
name = "aNEONSite/temperature")
## $Description
## [1] "Here is a description of the data"
##
## $Units
## [1] "Meters"
# look at the attributes of the aNEONsite group
h5readAttributes(file = "vegData.h5",
name = "aNEONSite")
## $Location
## [1] "Southern California"
##
## $SiteName
## [1] "San Joaquin Experimental Range"
# let's grab some data from the H5 file
testSubset <- h5read(file = "vegData.h5",
name = "aNEONSite/temperature")
testSubset2 <- h5read(file = "vegData.h5",
name = "aNEONSite/temperature",
index=list(NULL,1))
H5close()
Once we've extracted data from our H5 file, we can work with it
in R.
# create a quick plot of the data
hist(testSubset2)
### Challenge: Work with HDF5 Files
Time to practice the skills you've learned. Open up the D17_2013_SJER_vegStr.csv
in R.
Create a new HDF5 file called vegStructure.
Add a group in your HDF5 file called SJER.
Add the veg structure data to that folder.
Add some attributes the SJER group and to the data.
Now, repeat the above with the D17_2013_SOAP_vegStr csv.
Name your second group SOAP
Hint: read.csv() is a good way to read in .csv files.
Several factors contributed to the extreme flooding that occurred in Boulder,
Colorado in 2013. In this data activity, we explore and visualize the data for
stream discharge data collected by the United States Geological Survey (USGS).
The tutorial is part of the Data Activities that can be used
with the
Quantifying The Drivers and Impacts of Natural Disturbance Events Teaching Module.
Learning Objectives
After completing this tutorial, you will be able to:
Publish & share an interactive plot of the data using Plotly.
Things You'll Need To Complete This Lesson
Please be sure you have the most current version of R and, preferably,
RStudio to write your code.
R Libraries to Install:
ggplot2:install.packages("ggplot2")
plotly:install.packages("plotly")
Data to Download
We include directions on how to directly find and access the data from USGS's
National National Water Information System Database. However, depending on your
learning objectives you may prefer to use the
provided teaching data subset that can be downloaded from the NEON Data Skills account
on FigShare.
Set Working Directory This lesson assumes that you have set your working
directory to the location of the downloaded and unzipped data subsets.
R Script & Challenge Code: NEON data lessons often contain challenges that
reinforce learned skills. If available, the code for challenge solutions is found
in the downloadable R script of the entire lesson, available in the footer of each lesson page.
Research Question
What were the patterns of stream discharge prior to and during the 2013 flooding
events in Colorado?
About the Data - USGS Stream Discharge Data
The USGS has a distributed network of aquatic sensors located in streams across
the United States. This network monitors a suit of variables that are important
to stream morphology and health. One of the metrics that this sensor network
monitors is Stream Discharge, a metric which quantifies the volume of water
moving down a stream. Discharge is an ideal metric to quantify flow, which
increases significantly during a flood event.
As defined by USGS: Discharge is the volume of water moving down a stream or
river per unit of time, commonly expressed in cubic feet per second or gallons
per day. In general, river discharge is computed by multiplying the area of
water in a channel cross section by the average velocity of the water in that
cross section.
For more on stream discharge by USGS.
The USGS tracks stream discharge through time at locations across the United
States. Note the pattern observed in the plot above. The peak recorded discharge
value in 2013 was significantly larger than what was observed in other years.
Source: USGS, National Water Information System.
Obtain USGS Stream Gauge Data
This next section explains how to find and locate data through the USGS's
National Water Information System portal.
If you want to use the pre-compiled dataset at the FigShare link above, you can skip this
section and start again at the Work With Stream Gauge Data header.
Step 1: Search for the data
To search for stream gauge data in a particular area, we can use the
interactive map of all USGS stations.
By searching for locations around "Boulder, CO", we can find 3 gauges in the area.
For this lesson, we want data collected by USGS stream gauge 06730200 located on
Boulder Creek at North 75th St. This gauge is one of the few the was able to continuously
collect data throughout the 2013 Boulder floods.
You can directly access the data for this station through the "Access Data" link
on the map icon or searching for this site on the
National Water Information System portal .
On the
Boulder Creek stream gauge 06730200 page
, we can now see summary information about the types of data available for this
station. We want to select Daily Data and then the following parameters:
Available Parameters = 00060 Discharge (Mean)
Output format = Tab-separated
Begin Date = 1 October 1986
End Date = 31 December 2013
Now click "Go".
Step 2: Save data to .txt
The output is a plain text page that you must copy into a spreadsheet of
choice and save as a .csv. Note, you can also download the teaching dataset
(above) or access the data through an API (see Additional Resources, below).
Work with Stream Gauge Data
R Packages
We will use ggplot2 to efficiently plot our data and plotly to create interactive plots.
# load packages
library(ggplot2) # create efficient, professional plots
library(plotly) # create cool interactive plots
## Set your working directory to ensure R can find the file we wish to import and where we want to save our files. Be sure to move the downloaded files into your working directory!
wd <- "~/data/" # This will depend on your local environment
setwd(wd)
Import USGS Stream Discharge Data Into R
Now that we better understand the data that we are working with, let's import it into R. First, open up the discharge/06730200-discharge_daily_1986-2013.txt file in a text editor.
What do you notice about the structure of the file?
The first 24 lines are descriptive text and not actual data. Also notice that this file is separated by tabs, not commas. We will need to specify the
tab delimiter when we import our data.We will use the read.csv() function to import it into an R object.
When we use read.csv(), we need to define several attributes of the file
including:
The data are tab delimited. We will this tell R to use the "\\t"separator, which defines a tab delimited separation.
The first group of 24 lines in the file are not data; we will tell R to skip
those lines when it imports the data using skip=25.
Our data have a header, which is similar to column names in a spreadsheet. We
will tell R to set header=TRUE to ensure the headers are imported as column
names rather than data values.
Finally we will set stringsAsFactors = FALSE to ensure our data come in as individual values.
Let's import our data.
(Note: you can use the discharge/06730200-discharge_daily_1986-2013.csv file
and read it in directly using read.csv() function and then skip to the View
Data Structure section).
#import data
discharge <- read.csv(paste0(wd,"disturb-events-co13/discharge/06730200-discharge_daily_1986-2013.txt"),
sep= "\t", skip=24,
header=TRUE,
stringsAsFactors = FALSE)
#view first few lines
head(discharge)
## agency_cd site_no datetime X17663_00060_00003 X17663_00060_00003_cd
## 1 5s 15s 20d 14n 10s
## 2 USGS 06730200 1986-10-01 30 A
## 3 USGS 06730200 1986-10-02 30 A
## 4 USGS 06730200 1986-10-03 30 A
## 5 USGS 06730200 1986-10-04 30 A
## 6 USGS 06730200 1986-10-05 30 A
When we import these data, we can see that the first row of data is a second
header row rather than actual data. We can remove the second row of header
values by selecting all data beginning at row 2 and ending at the
total number or rows in the file and re-assigning it to the variable discharge. The nrow function will count the total
number of rows in the object.
# nrow: how many rows are in the R object
nrow(discharge)
## [1] 9955
# remove the first line from the data frame (which is a second list of headers)
# the code below selects all rows beginning at row 2 and ending at the total
# number of rows.
discharge <- discharge[2:nrow(discharge),]
Metadata
We now have an R object that includes only rows containing data values. Each
column also has a unique column name. However the column names may not be
descriptive enough to be useful - what is X17663_00060_00003?.
Reopen the discharge/06730200-discharge_daily_1986-2013.txt file in a text editor or browser. The text at
the top provides useful metadata about our data. On rows 10-12, we see that the
values in the 5th column of data are "Discharge, cubic feed per second (Mean)". Rows 14-16 tell us more about the 6th column of data,
the quality flags.
Now that we know what the columns are, let's rename column 5, which contains the
discharge value, as disValue and column 6 as qualFlag so it is more "human
readable" as we work with it
in R.
#view structure of data
str(discharge)
## 'data.frame': 9954 obs. of 5 variables:
## $ agency_cd: chr "USGS" "USGS" "USGS" "USGS" ...
## $ site_no : chr "06730200" "06730200" "06730200" "06730200" ...
## $ datetime : chr "1986-10-01" "1986-10-02" "1986-10-03" "1986-10-04" ...
## $ disValue : chr "30" "30" "30" "30" ...
## $ qualCode : chr "A" "A" "A" "A" ...
It appears as if the discharge value is a character (chr) class. This is
likely because we had an additional row in our data. Let's convert the discharge
column to a numeric class. In this case, we can reassign that column to be of
class: integer given there are no decimal places.
# view class of the disValue column
class(discharge$disValue)
## [1] "character"
# convert column to integer
discharge$disValue <- as.integer(discharge$disValue)
str(discharge)
## 'data.frame': 9954 obs. of 5 variables:
## $ agency_cd: chr "USGS" "USGS" "USGS" "USGS" ...
## $ site_no : chr "06730200" "06730200" "06730200" "06730200" ...
## $ datetime : chr "1986-10-01" "1986-10-02" "1986-10-03" "1986-10-04" ...
## $ disValue : int 30 30 30 30 30 30 30 30 30 31 ...
## $ qualCode : chr "A" "A" "A" "A" ...
Converting Time Stamps
We have converted our discharge data to an integer class. However, the time
stamp field, datetime is still a character class.
To work with and efficiently plot time series data, it is best to convert date
and/or time data to a date/time class. As we have both date and time date, we
will use the class POSIXct.
#view class
class(discharge$datetime)
## [1] "character"
#convert to date/time class - POSIX
discharge$datetime <- as.POSIXct(discharge$datetime, tz ="America/Denver")
#recheck data structure
str(discharge)
## 'data.frame': 9954 obs. of 5 variables:
## $ agency_cd: chr "USGS" "USGS" "USGS" "USGS" ...
## $ site_no : chr "06730200" "06730200" "06730200" "06730200" ...
## $ datetime : POSIXct, format: "1986-10-01" "1986-10-02" "1986-10-03" ...
## $ disValue : int 30 30 30 30 30 30 30 30 30 31 ...
## $ qualCode : chr "A" "A" "A" "A" ...
No Data Values
Next, let's query our data to check whether there are no data values in
it. The metadata associated with the data doesn't specify what the values would
be, NA or -9999 are common values
# check total number of NA values
sum(is.na(discharge$datetime))
## [1] 0
# check for "strange" values that could be an NA indicator
hist(discharge$disValue)
Excellent! The data contains no NoData values.
Plot The Data
Finally, we are ready to plot our data. We will use ggplot from the ggplot2
package to create our plot.
ggplot(discharge, aes(datetime, disValue)) +
geom_point() +
ggtitle("Stream Discharge (CFS) for Boulder Creek") +
xlab("Date") + ylab("Discharge (Cubic Feet per Second)")
Questions:
What patterns do you see in the data?
Why might there be an increase in discharge during a particular time of year?
Plot Data Time Subsets With ggplot
We can plot a subset of our data within ggplot() by specifying the start and
end times (in a limits object) for the x-axis with scale_x_datetime. Let's
plot data for the months directly around the Boulder flood: August 15 2013 -
October 15 2013.
# Define Start and end times for the subset as R objects that are the time class
start.end <- as.POSIXct(c("2013-08-15 00:00:00","2013-10-15 00:00:00"),tz= "America/Denver")
# plot the data - Aug 15-October 15
ggplot(discharge,
aes(datetime,disValue)) +
geom_point() +
scale_x_datetime(limits=start.end) +
xlab("Date") + ylab("Discharge (Cubic Feet per Second)") +
ggtitle("Stream Discharge (CFS) for Boulder Creek\nAugust 15 - October 15, 2013")
## Warning: Removed 9892 rows containing missing values (`geom_point()`).
We get a warning message because we are "ignoring" lots of the data in the
dataset.
Plotly - Interactive (and Online) Plots
We have now successfully created a plot. We can turn that plot into an interactive
plot using Plotly. Plotly
allows you to create interactive plots that can also be shared online. If
you are new to Plotly, view the companion mini-lesson
Interactive Data Vizualization with R and Plotly
to learn how to set up an account and access your username and API key.
Time subsets in plotly
To plot a subset of the total data we have to manually subset the data as the Plotly
package doesn't (yet?) recognize the limits method of subsetting.
Here we create a new R object with entries corresponding to just the dates we want and then plot that data.
# subset out some of the data - Aug 15 - October 15
discharge.aug.oct2013 <- subset(discharge,
datetime >= as.POSIXct('2013-08-15 00:00',
tz = "America/Denver") &
datetime <= as.POSIXct('2013-10-15 23:59',
tz = "America/Denver"))
# plot the data
disPlot.plotly <- ggplot(data=discharge.aug.oct2013,
aes(datetime,disValue)) +
geom_point(size=3) # makes the points larger than default
disPlot.plotly
# add title and labels
disPlot.plotly <- disPlot.plotly +
theme(axis.title.x = element_blank()) +
xlab("Time") + ylab("Stream Discharge (CFS)") +
ggtitle("Stream Discharge - Boulder Creek 2013")
disPlot.plotly
You can now display your interactive plot in R using the following command:
# view plotly plot in R
ggplotly(disPlot.plotly)
If you are satisfied with your plot you can now publish it to your Plotly account, if desired.
# set username
Sys.setenv("plotly_username"="yourUserNameHere")
# set user key
Sys.setenv("plotly_api_key"="yourUserKeyHere")
# publish plotly plot to your plotly online account if you want.
api_create(disPlot.plotly)
Additional Resources
Additional information on USGS streamflow measurements and data:
USGS data can be downloaded via an API using a command line interface. This is
particularly useful if you want to request data from multiple sites or build the
data request into a script.
Read more here about API downloads of USGS data.
R Script & Challenge Code: NEON data lessons often contain challenges that
reinforce learned skills. If available, the code for challenge solutions is found
in the downloadable R script of the entire lesson, available in the footer of each lesson page.
Research Question: How do We Measure Changes in Terrain?
Questions
How can LiDAR data be collected?
How might we use LiDAR to help study the 2013 Colorado Floods?
Additional LiDAR Background Materials
This data activity below assumes basic understanding of remote sensing and
associated landscape models and the use of raster data and plotting raster objects
in R. Consider using these other resources if you wish to gain more background
in these areas.
Digital Terrain Models, Digital Surface Models and Canopy Height
Models are three common LiDAR-derived data products. The digital terrain model
allows scientists to study changes in terrain (topography) over time.
How might we use a CHM, DSM or DTM model to better understand what happened
in the 2013 Colorado Flood?
Would you use only one of the models or could you use two or more of them
together?
In this Data Activity, we will be using Digital Terrain Models (DTMs).
More Details on LiDAR
If you are particularly interested in how LiDAR works consider taking a closer
look at how the data are collected and represented by going through this tutorial
on
"Light Detection and Ranging."
Digital Terrain Models
We can use a digital terrain model (DTM) to view the surface of the earth. By
comparing a DTM from before a disturbance event with one from after a disturbance
event, we can get measurements of where the landscape changed.
First, we need to load the necessary R packages to work with raster files and
set our working directory to the location of our data.
Then we can read in two DTMs. The first DTM preDTM3.tif is a terrain model created from data
collected 26-27 June 2013 and the postDTM3.tif is a terrain model made from data collected
on 8 October 2013.
Among the information we now about our data from looking at the raster structure,
is that the units are in meters for both rasters.
Hillshade layers are models created to add visual depth to maps. It represents
what the terrain would look like in shadow with the sun at a specific azimuth.
The default azimuth for many hillshades is 315 degrees -- to the NW.
# Creating hillshade for DTM_pre & DTM_post
# In order to generate the hillshde, we need both the slope and the aspect of
# the extent we are working on.
DTM_pre_slope <- terrain(DTM_pre, v="slope", unit="radians")
DTM_pre_aspect <- terrain(DTM_pre, v="aspect", unit="radians")
DTM_pre_hillshade <- shade(DTM_pre_slope, DTM_pre_aspect)
DTM_post_slope <- terrain(DTM_post, v="slope", unit="radians")
DTM_post_aspect <- terrain(DTM_post, v="aspect", unit="radians")
DTM_post_hillshade <- shade(DTM_post_slope, DTM_post_aspect)
Now we can plot the raster objects (DTM & hillshade) together by using add=TRUE
when plotting the second plot. To be able to see the first (hillshade) plot,
through the second (DTM) plot, we also set a value between 0 (transparent) and 1
(not transparent) for the alpha= argument.
# plot Pre-flood w/ hillshade
plot(DTM_pre_hillshade,
col=grey(1:90/100), # create a color ramp of grey colors for hillshade
legend=FALSE, # no legend, we don't care about the values of the hillshade
main="Pre-Flood DEM: Four Mile Canyon, Boulder County",
axes=FALSE) # makes for a cleaner plot, if the coordinates aren't necessary
plot(DTM_pre,
axes=FALSE,
alpha=0.3, # sets how transparent the object will be (0=transparent, 1=not transparent)
add=TRUE) # add=TRUE (or T), add plot to the previous plotting frame
Zoom in on the main stream bed. Are changes more visible? Can you tell
where erosion has occurred? Where soil deposition has occurred?
Digital Elevation Model of Difference (DoD)
A Digital Elevation Model of Difference (DoD) is a model of the
change (or difference) between two other digital elevation models - in our case
DTMs.
# DoD: erosion to be neg, deposition to be positive, therefore post - pre
DoD <- DTM_post-DTM_pre
plot(DoD,
main="Digital Elevation Model (DEM) Difference",
axes=FALSE)
Here we have our DoD, but it is a bit hard to read. What does the scale bar tell
us?
Everything in the yellow shades are close to 0m of elevation change, those areas
toward green are up to 10m increase of elevation, and those areas to red and
white are a 5m or more decrease in elevation.
We can see a distribution of the values better by viewing a histogram of all
the values in the DoD raster object.
# histogram of values in DoD
hist(DoD)
## Warning: [hist] a sample of25% of the cells was used
Most of the areas have a very small elevation change. To make the map easier to
read, we can do two things.
Set breaks for where we want the color to represent: The plot of the DoD
above uses a continuous scale to show the gradation between the loss of
elevation and the gain in elevation. While this provides a great deal of
information, in this case with much of the change around 0 and only a few outlying
values near -5m or 10m a categorical scale could help us visualize the data better.
In the plotting code we can set this with the breaks= argument in the plot()
function. Let's use breaks of -5, -1, -0.5, 0.5, 1, 10 -- which will give use 5
categories.
Change the color scheme: We can specify a color for each of elevation
categories we just specified with the breaks.
ColorBrewer 2.0 is a great reference for choosing mapping color palettes and
provide the hex codes we need for specifying the colors of the map. Once we've
chosen appropriate colors, we can create a vector of those colors and then use
that vector with the `col=` argument in the `plot()` function to specify these.
Let's now implement these two changes in our code.
# Color palette for 5 categories
difCol5 = c("#d7191c","#fdae61","#ffffbf","#abd9e9","#2c7bb6")
# Alternate palette for 7 categories - try it out!
#difCol7 = c("#d73027","#fc8d59","#fee090","#ffffbf","#e0f3f8","#91bfdb","#4575b4")
# plot hillshade first
plot(DTM_post_hillshade,
col=grey(1:90/100), # create a color ramp of grey colors
legend=FALSE,
main="Elevation Change Post-Flood: Four Mile Canyon, Boulder County",
axes=FALSE)
# add the DoD to it with specified breaks & colors
plot(DoD,
breaks = c(-5,-1,-0.5,0.5,1,10),
col= difCol5,
axes=FALSE,
alpha=0.3,
add =T)
Question
Do you think this is the best color scheme or set point for the breaks? Create
a new map that uses different colors and/or breaks. Does it more clearly show
patterns than this plot?
Optional Extension: Crop to Defined Area
If we want to crop the map to a smaller area, say the mouth of the canyon
where most of the erosion and deposition appears to have occurred, we can crop
by using known geographic locations (in the same CRS as the raster object) or
by manually drawing a box.
Method 1: Manually draw cropbox
# plot the rasters you want to crop from
plot(DTM_post_hillshade,
col=grey(1:90/100), # create a color ramp of grey colors
legend=FALSE,
main="Pre-Flood Elevation: Four Mile Canyon, Boulder County",
axes=FALSE)
plot(DoD,
breaks = c(-5,-1,-0.5,0.5,1,10),
col= difCol5,
axes=FALSE,
alpha=0.3,
add =T)
# crop by designating two opposite corners
cropbox1 <- draw()
After executing the draw() function, we now physically click on the plot
at the two opposite corners of the box you want to crop to. You should see a
red bordered polygon display on the raster at this point.
When we call this new object, we can view the new extent.
# view the extent of the cropbox1
cropbox1
## [1] 473814 474982 4434537 4435390
It is a good idea to write this new extent down, so that you can use the extent
again the next time you run the script.
Method 2: Define the cropbox
If you know the desired extent of the object you can also use it to crop the box,
by creating an object that is a vector containing the four vertices (x min,
x max, y min, and y max) of the polygon.
# desired coordinates of the box
cropbox2 <- c(473792.6,474999,4434526,4435453)
Once you have the crop box defined, either by manually clicking or by setting
the coordinates, you can crop the desired layer to the crop box.
# crop desired layers to the cropbox2 extent
DTM_pre_crop <- crop(DTM_pre, cropbox2)
DTM_post_crop <- crop(DTM_post, cropbox2)
DTMpre_hill_crop <- crop(DTM_pre_hillshade,cropbox2)
DTMpost_hill_crop <- crop(DTM_post_hillshade,cropbox2)
DoD_crop <- crop(DoD, cropbox2)
# plot the pre- and post-flood elevation + DEM difference rasters again, using the cropped layers
# PRE-FLOOD (w/ hillshade)
plot(DTMpre_hill_crop,
col=grey(1:90/100), # create a color ramp of grey colors:
legend=FALSE,
main="Pre-Flood Elevation: Four Mile Canyon, Boulder County ",
axes=FALSE)
plot(DTM_pre_crop,
axes=FALSE,
alpha=0.3,
add=T)
# POST-FLOOD (w/ hillshade)
plot(DTMpost_hill_crop,
col=grey(1:90/100), # create a color ramp of grey colors
legend=FALSE,
main="Post-Flood Elevation: Four Mile Canyon, Boulder County",
axes=FALSE)
plot(DTM_post_crop,
axes=FALSE,
alpha=0.3,
add=T)
# ELEVATION CHANGE - DEM Difference
plot(DTMpost_hill_crop,
col=grey(1:90/100), # create a color ramp of grey colors
legend=FALSE,
main="Post-Flood Elevation Change: Four Mile Canyon, Boulder County",
axes=FALSE)
plot(DoD_crop,
breaks = c(-5,-1,-0.5,0.5,1,10),
col= difCol5,
axes=FALSE,
alpha=0.3,
add =T)
Now you have a graphic of your particular area of interest.
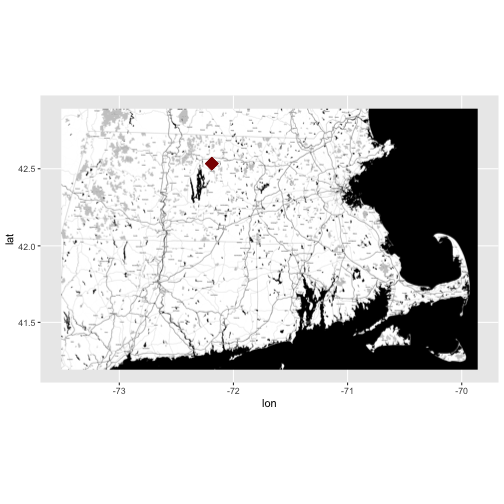
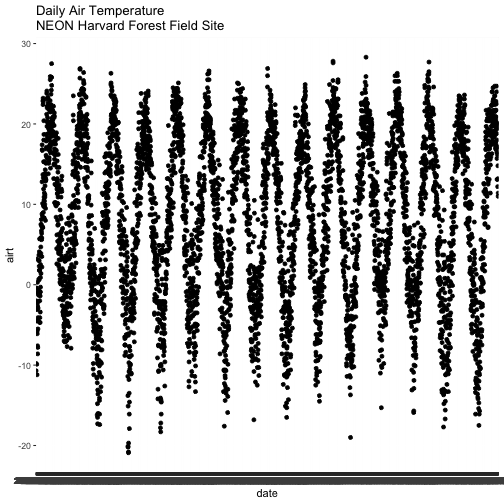
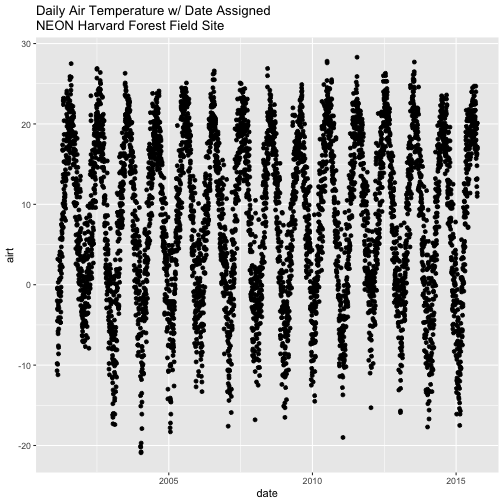
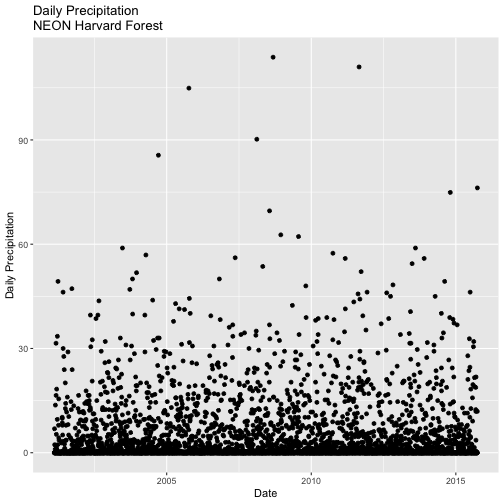
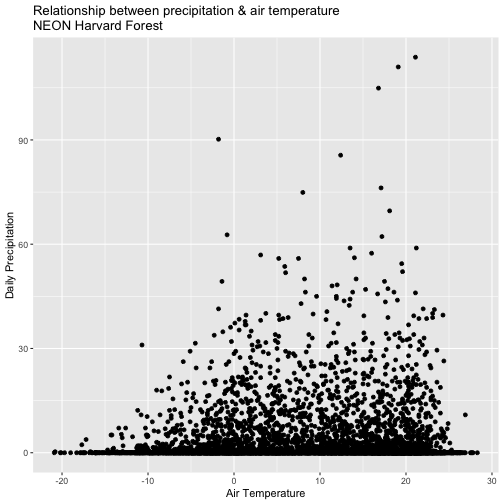
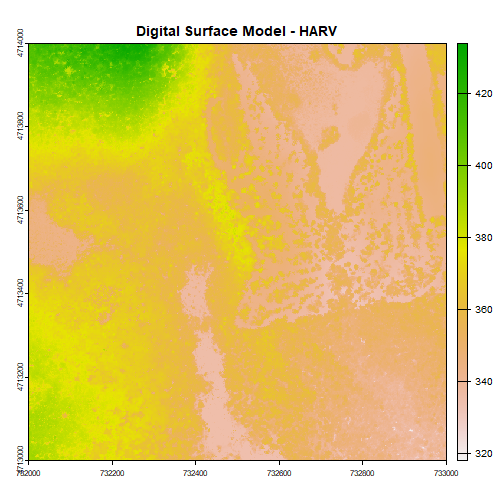
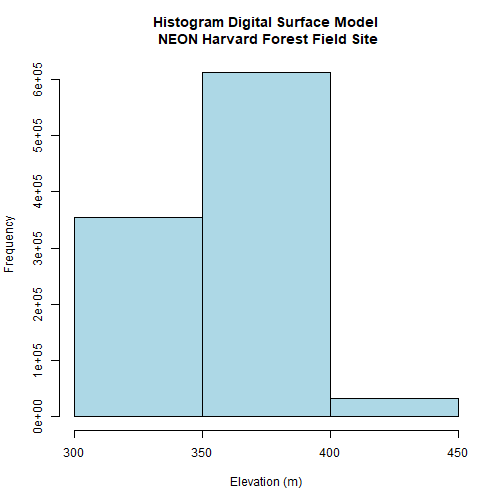
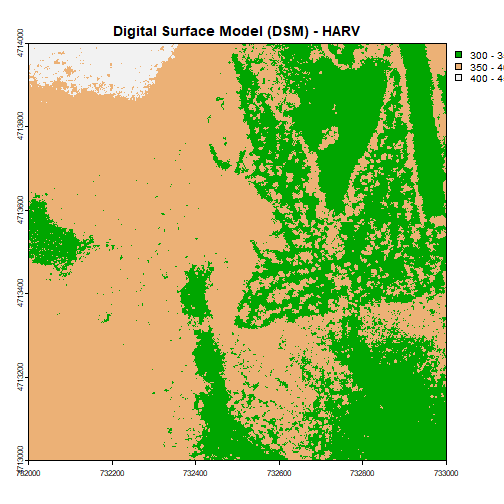
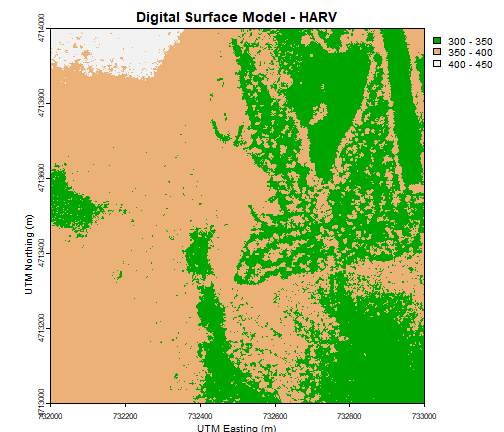
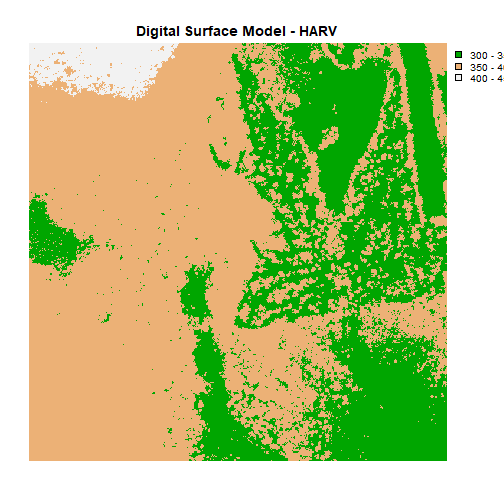
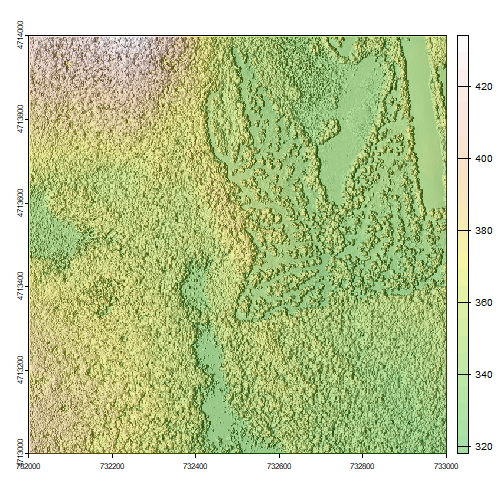
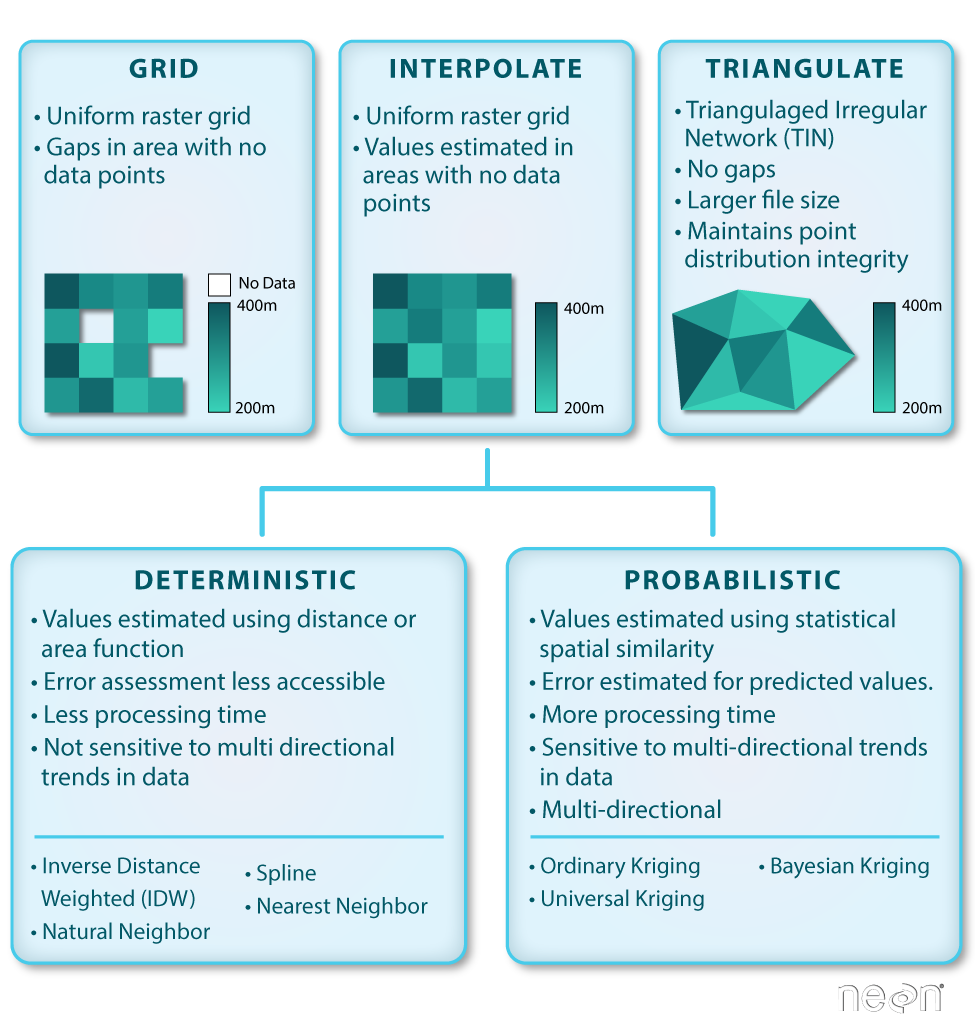



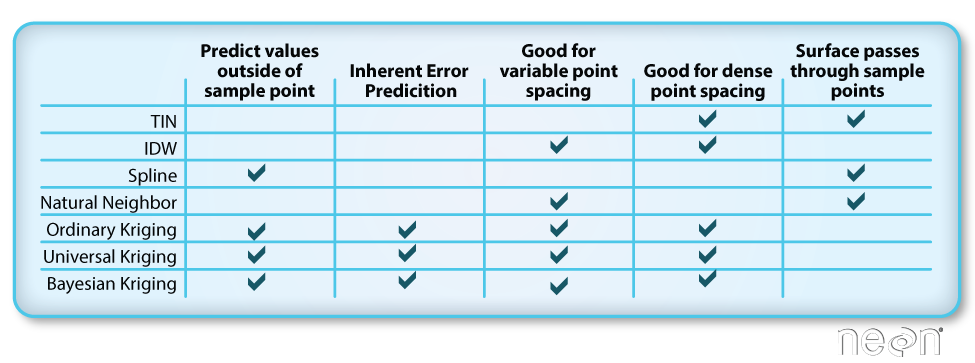
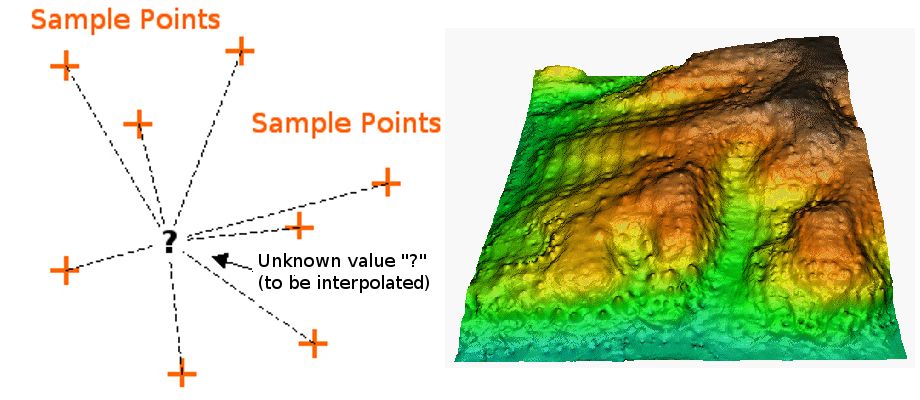
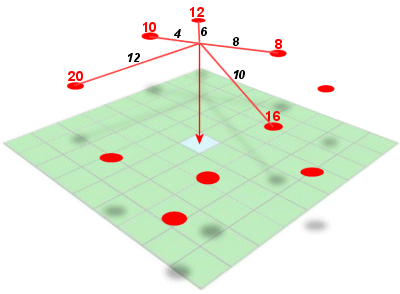



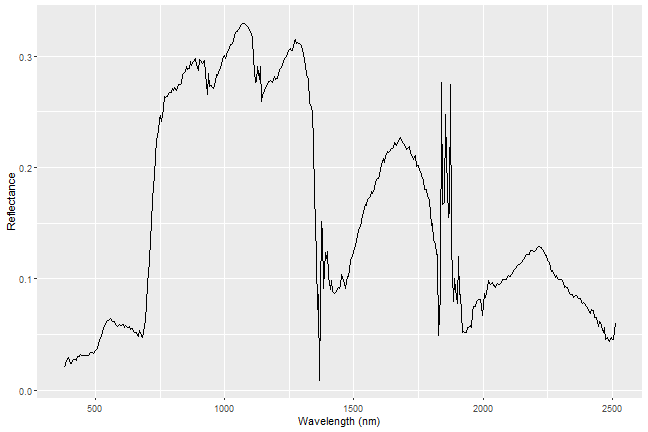
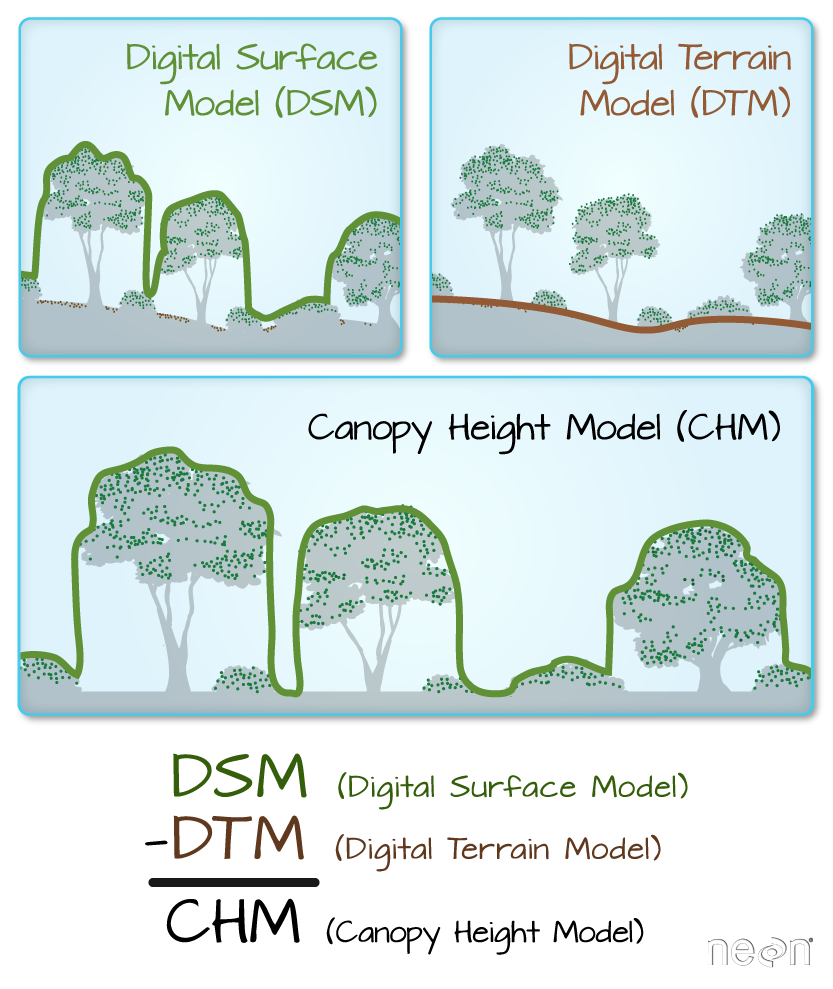


.")
.")


.")